See how thermodynamics can be used to improve your lead optimization process. Renin is an aspartyl protease involved in the production of angiotensin II, a potent vasoconstrictor. Renin inhibitors can prevent blood vessel constriction and could therefore be useful for the treatment of hypertension. High throughput screening efforts identified a small molecule renin inhibitor with a core substituted diaminopyrimidine ring. Parallel medicinal chemistry efforts based on this lead resulted in compound 1. A complex of 1 bound to renin was crystallized and structural data obtained by X-ray diffraction. The structure indicated there were adjacent unoccupied binding pockets. Synthetic efforts were initiated to extend functionality into these pockets to improve affinity and adjust pharmacokinetic parameters.
Thermodynamics data for inhibitor binding to renin were also collected using isothermal titration calorimetry. These data were used to help guide inhibitor optimization by suggesting molecular alterations to improve binding affinity from both a thermodynamic and structural perspective. Addition of a methoxy-propyl group extending into the S3 subpocket improved inhibitor affinity and resulted in greater binding enthalpy. Initial additions to the pyrimidine ring template that extended into the large hydrophobic S2 pocket did not improve affinity and dramatically altered the thermodynamic driving force for the binding interaction. Binding of the core template was enthalpically driven whereas binding of initial inhibitors with S2 extensions were both enthalpically and entropically driven but lost significant binding enthalpy. Additional electrostatic interactions were then incorporated in the S2 extension to improve binding enthalpy while taking advantage of the favorable entropy.
*Abbreviations used: NHANES, National Health and Nutrition Examination Survey; ACE, angiotensin-converting enzyme; ARB, angiotensin II receptor blocker; CHO, Chinese hamster ovary; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; DMSO, dimethyl sulfoxide; MWCO, molecular weight cutoff; TPCK trypsin, N-tosyl-Lphenylalanine chloro-methyl ketone-trypsin; PEG, polyethylene glycol; ITC, isothermal titration calorimetry; S3sp, S3 subpocket; VDW, van der Waals.
Analysis of data from the 1999-2002 National Health and Nutrition Examination Survey (NHANES)* indicated that an estimated 65 million individuals in the United States have hypertension.1 This analysis also determined that only 63.4% of the individuals sampled knew they were hypertensive, 45.3% were being treated, 29.3% had their blood pressure under control, but 70.7% of the total hypertensive population (~46 million people) did not1. Comparison of this data to earlier data indicated that since 1960 there have been improvements in awareness, treatment, and control of hypertension,2 but there is substantial room to improve both the identification of individuals at risk for hypertension and the treatment options. Current treatment options for hypertension range from lifestyle modifications, including lowering salt intake and reducing weight, to pharmaceutical intervention. Pharmaceutical therapies currently available include diuretics, alpha-blockers, alpha-beta-blockers, beta-blockers, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin II receptor blockers (ARB).3
ACE inhibitors reduce blood pressure by inhibiting production of the potent vasoconstrictor angiotensin II and ARB inhibitors block binding of angiotensin II to the angiotensin receptor. Angiotensin II is produced in the final step of a two-step enzymatic cleavage of the glycoprotein angiotensinogen. In the first step, renin, an aspartyl protease, cleaves the decapeptide angiotensin I, from angiotensinogen.
Then, ACE catalyzes a two residue cleavage of angiotensin I to form the octapeptide angiotensin II. Binding of angiotensin II to the angiotensin receptor initiates a cascade of events including vasoconstriction, and retention of sodium and water that can lead to hypertension. Inhibition of angiotensin II production thereby provides a rational opportunity for therapeutic treatment of hypertension. Several marketed medications inhibit production of angiotensin II through ACE inhibition and their clinical benefits have been reviewed.4 Except for dry cough reported in some treated patients,5 ACE inhibitors have been widely accepted based on good efficacy and safety profiles. While ACE inhibitors and many of the existing therapies are effective alone or in-combination, as previously mentioned, there remains a portion of hypertensive patients that do not attain the desired reduction in blood pressure or do not continue treatment due to undesired treatment effects.
In addition, some hypertensive patients do not respond to a combination of treatments6 and opportunities remain for improved treatment options.
Since ACE catalyzes the final step in the metabolic pathway of angiotensin II, inhibitors involved further up-stream in the metabolic pathway, such as renin inhibitors, have been sought to help reduce unwanted side effects. Many mechanistic studies of renin have been performed and the high-resolution structure of renin bound to substrate is known. Despite the wealth of information, developing functional nonpeptidic inhibitors of renin has proven challenging. Initial renin inhibitors were peptidic and suffered from rapid metabolic clearance and short plasma half-life. More recently some nonpeptidic inhibitors have been identified and, the renin inhibitor aliskiren, has completed Phase III clinical studies and has been submitted to the FDA for review.7
A high throughput screen for inhibitors of renin was performed at Pfizer that resulted in the identification of a unique nonpeptidic small molecule inhibitor with a diaminopyrimidine core. A round of parallel chemistry resulted in compound 1 that inhibited 50% of renin activity (IC50) in-vitro at a compound concentration of 6 μM. This was an improvement over the IC50 of 27 μM for the initial lead but increased potency was still required to be a viable drug candidate. Additional chemical modifications were done to the pyrimidine core to further optimize the lead and improve affinity for the substrate binding pocket.8-10 Many of the synthesized molecules were complexed with renin and crystallized. Detailed structural information on the binding of the lead to renin was obtained from X-ray crystallographic experiments. In addition, thermodynamics for the binding interactions were measured to determine the enthalpic and entropic contributions to binding affinity. Structural and thermodynamic information were then used in tandem to provide insights into the structure activity relationship for ligand interactions with the binding pockets. This combination of techniques was useful in building a unique nonpeptidic small molecule inhibitor of renin.
Improved sensitivity, reliability, and ease of use of commercial micro-calorimeters11 has increased the use of thermodynamic information in inhibitor design.
For example, thermodynamic evaluation of ligand binding interactions has been successfully used in the design of improved inhibitors of HIV protease.12;13 These studies showed that first generation protease inhibitors were largely entropically driven and lost significant inhibitory activity with mutations in the HIV protease. Subsequent generation inhibitors with enthalpically driven binding retained significant HIV protease inhibitory activity upon enzyme mutations. Enthalpically driven inhibitors were more flexible and able to accommodate protease mutations that left the sterically constrained first-generation inhibitors inactive.
Human preprorenin was cloned from Human Fetal Kidney Quick-Clone cDNA (Clontech, 7170-1). Preprorenin was cloned into pcDNA3.1(+) (Invitrogen, V790-20) at the BamHI and XbaI restriction sites. The clone was confirmed by sequencing and compared with the published human preprorenin sequence (Accession E01074).
Transfection of Chinese hamster ovary (CHO) cells, cell type K1 (cat. no. CCL-61, American Type Culture Collection), was performed by using Effectene transfection reagent (cat. no. 301425, Qiagen). CHO cells, at 70% confluency, in a 150mm tissue culture dish (cat. no. 3025, Falcon) were used for each transfection. A standard transfection protocol employed 4 μg renin plasmid DNA and 120 μL Effectene reagent in a final volume of 20 mL of media. In addition, 0.4 μg of pcDNA6 V5/HisB plasmid was transfected with the renin plasmid so as to impart Blasticidin resistance. The transfection mixture remained on the cells overnight (~20 hrs).
To establish a stable line, the transfected CHO cells were grown in Dulbecco’s modified Eagle’s medium (DMEM)-F12 selection media containing 500 μg/mL geneticin and 6 μg/mL blasticidin. Following approximately 10 days of selection pressure, the surviving cells formed distinct colonies. For clonal selection, flasks of cells were counted and then diluted to allow for plating of 1 cell/well using a 96-well plate format. Microscopic examination was used to determine wells containing single colonies. Expansion of each colony continued for about 7 days in a 96-well format. Each colony was expanded into subsequently larger wells. For long-term storage, cells were cryopreserved in 95% fetal bovine serum (FBS, cat. no.16000-036, Invitrogen) and 5% dimethyl sulfoxide (DMSO) following a standard procedure.
CHO cells expressing preprorenin were grown both batch wise and in perfusion mode in a basket bioreactor using serum free medium. Batch runs were seeded at 1 x 105/ml and grown at 37°C for 6-7 days, reaching a density of about 1.5 x 106/ml.
Perfusion runs (3.5L bioreactor) were seeded with approximately 1 x 109 cells, which were allowed to attach to polyester fiber disks contained in a basket within the bioreactor. The cells were grown at 37°C and reached a maximum density of 1.5 x 1011 as estimated from glucose utilization. Fresh medium was perfused beginning at 72 hours at a rate of 2 L per day and gradually increased to a maximum of 20 L per day, with preprorenin being expressed for at least 38 days. The resulting secreted prorenin was glycosylated as indicated by mass spectrometry but the glycosylation state was not directly compared to native human prorenin.
Purification was modified from [Ref14]. All steps were performed at 4°C unless otherwise noted. CHO media containing recombinant human prorenin was concentrated 150-fold using 10-kDa molecular weight cutoff (MWCO) ultrafiltration, and 40% saturated ammonium sulfate was added. The slurry was stirred overnight, and insoluble material was removed by centrifugation at 16,000g. The supernatant was buffer exchanged using 10-kDa MWCO ultrafiltration and loaded onto Blue Sepharose 6 Fast Flow (cat. no. 17-0948-02, GE Healthcare) in 20 mM Tris–HCl (pH 8.0) and 0.2 MNaCl. Prorenin was eluted with a linear gradient to 1.4 M NaCl and was buffer exchanged versus 20 mM Tris–HCl (pH 7.4) and 0.1 M NaCl. Prorenin was mixed with immobilized N-tosyl-L-phenylalanine chloromethyl ketone-trypsin (TPCK trypsin, cat. no. 20230, Pierce Biotechnology,) for 3 h at 200C. Mature renin was eluted and loaded onto Blue Sepharose 6 Fast Flow in 20 mM Tris–HCl (pH 7.4) and 0.1 M NaCl. The unbound fraction containing renin was buffer exchanged and loaded onto POROS S/20 (cat. no. 1-3022, Applied Biosystems) in 10 mM sodium acetate (pH 5.0) and 5.0 mM NaCl. Renin was eluted with a linear gradient to 1.0 M NaCl and was dialyzed versus 20 mM Tris–HCl (pH 7.0) and 0.1 MNaCl. Purified renin was concentrated to 12 mg ml-1 and stored at -70°C. Final yield was typically 0.2-0.4 mg crystallizable renin per liter of prorenin-containing media.
Purified renin was crystallized by the vapor-diffusion in hanging drop method. Equal volumes (2 μL) of mature renin (8-12 mg mL-1) and reservoir solution (10–20% polyethylene glycol [PEG] 3350, 50 mM sodium citrate [pH 4.5], and 0.6
|
M NaCl) were mixed and equilibrated at 20°C.15 The crystals belong to the cubic P213 space group with the unit cell dimension of approximately 141 Å. Suitable crystals (~0.25 mm on each face) were transferred into a fresh 4 μL hanging drop containing 2.5 mM compound dissolved in well solution with an additional 2-3% PEG 3350. The drop was incubated at 20°C, typically for 48 hours. The crystals were cryoprotected by transferring to a 4 μL drop of 20% ethylene glycol in well solution, and then immersed in liquid nitrogen.
X-ray diffraction data of the renin complexes inhibited with compounds 1, 3 and 11 (Table 1) were collected at -180°C at the Advanced Photon Source facility on beamline 17-ID operated by the Industrial Macromolecular Crystallography Association. The crystals of compound 1 and 11 scattered X-rays to 1.9 and 2.25 Å resolution, respectively. The diffraction pattern from the crystal of compound 3 was considerably weaker, 2.6 Å resolution. Auto-indexing and processing of the measured intensity data were carried out with the HKL2000 software package. 16 The intensity data collection statistics are summarized in Table 2.
| Compound 1 | Compound 3 | Compound 11 | |
|---|---|---|---|
| Space group | P213 | P213 | P213 |
| Cell constantsa=b=c (Å) | 140.86 | 141.16 | 141.53 |
| α=β=γ (deg.) | 90.0 | 90.0 | 90.0 |
| Complexes/asymmetric unit | 2 | 2 | 2 |
| Resolution (Å) | 1.90 | 2.60 | 2.24 |
| Outmost range (Å) | 1.97-1.90 | 2.69-2.60 | 2.32-2.24 |
| Observations | 216,560 | 120,225 | 333,042 |
| Unique reflections | 70,222 | 28,798 | 45,325 |
| Outmost range | 6,373 | 2,858 | 4,278 |
| Redundancy | 3.1 | 4.2 | 7.3 |
| Rsym | 0.063 | 0.125 | 0.073 |
| Outmost range | 0.606 | 0.539 | 0.627 |
| Completeness (%) | 96 | 99 | 99 |
| Outmost range (Å) | 88 | 99 | 94 |
| I/sigma (I) | 16.98 | 11.86 | 31.48 |
| Outmost range (Å) | 1.01 | 2.08 | 3.28 |
The crystal structure of renin complexed with compound 1 was determined by molecular replacement using renin coordinates of the previously determined renin structure (PDB code 2I4Q) as an initial model. The rotation and translation searches were carried out with the MOLREP program,17 Collaborative Computational Project, Number 4. The rotation search provided two outstanding solutions with the Rfree/σ values of 8.54 and 5.68, corresponding to each of two molecules in the asymmetric unit related by a non-crystallographic 2 fold rotation axis. The position of one molecule was determined with a translation function that provided a R-value of 0.540 (score of 0.60); the second molecule improved the R-value to 0.482 (score of 0.704). The coordinates from the molecular replacement solution were further optimized by rigid-body refinement followed by slow-cooling simulated annealing and individual B-factor refinement using CNX 2002 (Accelrys).18 This decreased the Rwork and Rfree values to 0.194/0.229. Electron (2Fo-Fc) and difference (Fo-Fc) density maps were utilized for interactive fitting of protein structures into electron density using Coot19 and x-BUILD.20 Protein model building was alternated with coordinate minimization and individual B-factor refinement using Refmac 5.0.21 The parts of the protein structure that underwent conformational changes in respect to the initial model were fixed based on an analysis of the electron and difference density maps after deletion and then reconstruction. The calculated (2Fo-Fc) electron and (Fo-Fc) difference density maps showed well-defined electron density corresponding to the active site inhibitor, which was omitted from the calculations (Figure 1B). Placement of compound 1 to electron density was carried out with X-LIGAND (Accelrys).22 Solvent water molecules were added periodically based on examination of difference density maps using Coot and X-SOLVATE. Final protein coordinates were validated using PROCHECK.23
|
|
Crystal structures of renin complexed with compounds 3 and 11 were determined by the molecular replacement method. Renin coordinates from the refined structure of compound 1 were used as the starting model. Protocols and crystallographic software packages used in structure refinement and electron density fitting of complexes 3 and 11 were identical to those used in structure determination of renin complexed with compound 1. The OMIT (Fo-Fc) electron density map for compounds 3 and 11 are shown in Figures 3B and 4B. The final refinement statistics are presented in Table 3.
| Compound 1 | Compound 3 | Compound 11 | ||
|---|---|---|---|---|
| Restraints | Target | RMSΔ | RMSΔ | RMSΔ |
| Distance (Å) | ||||
| Bond distances | 0.020 | 0.009 | 0.011 | 0.012 |
| Bond angles | 2.00 | 1.276 | 1.361 | 1.411 |
| Chiral centers | 0.200 | 0.114 | 0.076 | 0.085 |
| Planar groups | 0.020 | 0.004 | 0.004 | 0.005 |
| Non-bonded contacts (Å) | ||||
| VDW repulsions | 0.200 | 0.1293 | 0.202 | 0.192 |
| VDW torsion | 0.200 | 0.179 | 0.185 | 0.179 |
| H-Bond | 0.200 | 0.142 | 0.146 | 0.129 |
| Isotropic thermal factors (Å2) | ||||
| Main-chain bond | 1.5 | 0.5 | 0.7 | 0.8 |
| Main-chain angle | 2.0 | 1.0 | 0.8 | 1.0 |
| Side-chain bond | 3.0 | 1.4 | 1.2 | 1.5 |
| Side-chain angle | 4.5 | 2.1 | 1.8 | 2.3 |
| R -factor | 0.194 | 0.212 | 0.206 | |
| Rfreea | 0.229 | 0.256 | 0.242 | |
| B-value of chains A/B (Å2) | 36.5/41.4 | 42.2/46.0 | 46.1/50.0 | |
| B-value of ligands A/B (Å2) | 28.2 | 34.1/47.2 | 33.7/37.9 | |
| B-value of solvent A/B (Å2) | 39.6 | 37.9 | 41.9 | |
| No. of protein residues | 670 | 674 | 674 | |
| No. of water molecules | 408 | 85 | 242 | |
| PDB code | 2IKO | 2IKU | 2IL2 |
a R free is calculated for the 5% of the data that were withheld from refinement
The coordinates of the structures have been deposited in the RCSB Protein Data Bank (compound 1, access PDB code: 2IKO; compound 3, access PDB code: 2IKU; compound 11, access PDB code: 2IL2).
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal VP isothermal titrating microcalorimeter. Data collection, analysis and plotting were performed using a Windows-based software package (Origin, version 7.0) supplied by MicroCal. The titrating microcalorimeter consisted of a sample and reference cell held in an adiabatic enclosure. The isothermal titration calorimeter was calibrated by comparing the measured areas of applied heat pulses to known values. Known and experimentally measured values agreed to within 2%. To minimize heat of dilution effects resulting from differences in buffer composition between ligand and protein, ligands were dissolved in dialysate buffer from the final step in the renin purification. Ligand and protein solutions were degassed prior to analysis. The reference cell was filled with dialysate buffer. Since aqueous solubilities of ligands was poor, buffered renin at 300 μM was placed in a 250 μL syringe and 20 μM ligand was placed in the 1.375 mL sample cell. The concentration of renin was determined using an A280 of 1.06 for a 1 mg/mL solution. Typically, 30 injections, 4 μL per injection, of renin were made by a computer controlled injector into the sample cell filled with ligand solution. The syringe stir rate was 300 rpm. Heat adsorbed or released with each injection was measured by the calorimeter. Titration isotherms for the binding interactions were comprised of the differential heat flow for each injection. These were integrated to provide the enthalpy change with each injection. Heats of dilution obtained by injecting renin into final purification buffer were insignificant. Isotherms fit well to a single-site model using an iterative non-linear least-squares algorithm.11 All parameters were floated during the iterations.
Binding isotherms fit by this method provided the equilibrium association or binding constant, (Ka), the change in enthalpy, (ΔH), and stoichiometry of binding, (N). Binding stoichiometry was 1:1 within experimental error. The change in free energy (ΔG) and change in entropy (ΔS) were then determined using the following equation:
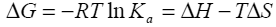
|
Equation 1 |
where R is the universal gas constant, T is the temperature in degrees Kelvin, and other parameters are as previously defined.
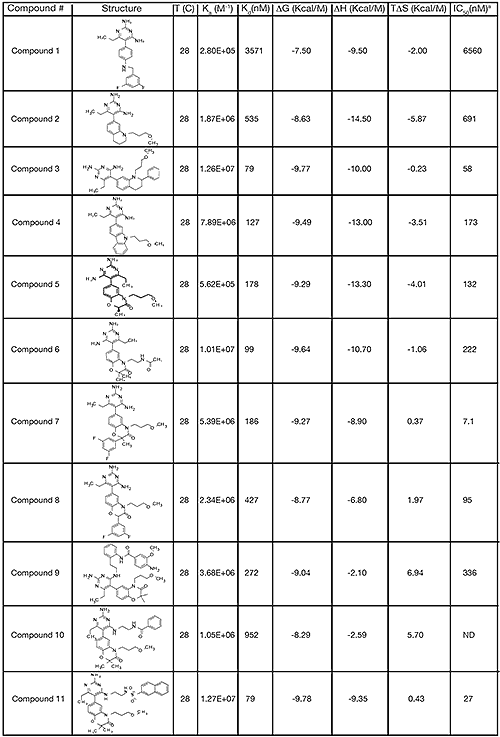
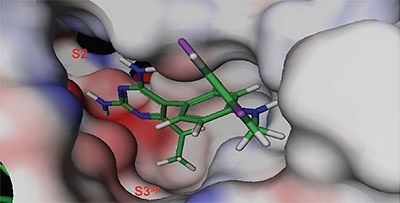
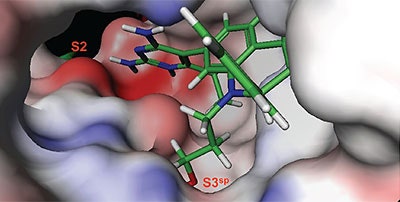
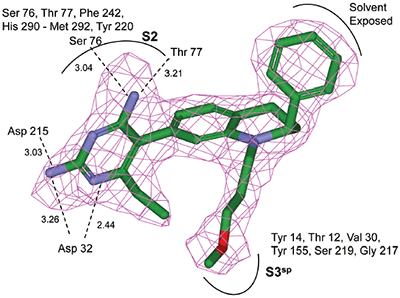
The structure and thermodynamic binding parameters of a small molecule renin inhibitor compound 1 are provided in Table 1. IC50 values reported in Table 1 were determined in duplicate using a previously described fluorescence tGFP assay.24 This 3,5-difluoro analog contains the core diaminopyrimidine ring system of the lead identified from high throughput screening. Gibbs free energy of binding, ΔG= -7.5 kcal/M, for 1 was the weakest of the compounds listed as calculated from the equilibrium dissociation constant, (Kd), of 3.6 μM determined by ITC. Weak affinity of the lead was consistent with weak renin inhibition of the compound (IC50 = 6.6 μM). The active site co-structure of 1 bound to renin is shown in Figure 1 as determined from X-ray crystallography to a resolution of 1.9 Å. Several hydrogen bonds were formed between the diaminopyrimidine and renin including H-bonds from the 2-amino group to Thr77 and Ser76, an H-bond between Asp215 and the 4-amino group, an H-bond between Asp32 and the 4-amino group, and an H-bond between Asp32 and the pyrimidine ring nitrogen at position 5. This strong network of H-bonds contributed to the significant binding enthalpy of –9.5 kcal/M for 1. The large hydrophobic S2 (residue numbers, Ser76, Thr77, Phe242, His290- Met292, and Tyr220) and smaller S3 subpocket (S3sp) (residue numbers Val 30, Tyr 14, Thr 12, Tyr 155, Ser 219, and Gly 217) were unoccupied by 1 (Figure 1A). Alterations to the core diaminopyrimidine template were then explored to take advantage of these nearby binding pockets to improve affinity and pharmacokinetic parameters of the lead.
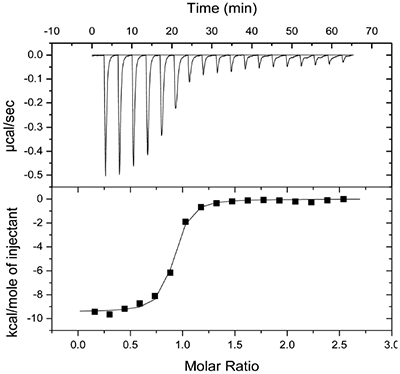
Molecular modeling studies indicated that an appropriately positioned methoxypropyl group could be extended into the S3sp by attaching it to a tetrahydroquinoline ring tethered to the diaminopyrimidine ring of the lead. Compound 2, shown in Table 1, was synthesized based on modeling results [9]. Extension of the methoxypropyl of 2 into S3sp significantly improved ligand affinity as indicated by the thermodynamic parameters listed in Table 1. The equilibrium dissociation constant (Kd) or binding constant was 530 nM and the change in binding enthalpy was –14.5 kcal/M. Improved free binding energy resulted from a ΔΔH of –5 kcal/M compared to 1. Improved affinity also translated into improved inhibition of enzyme activity, renin IC50 = 691 nM, a 10-fold increase compared to 1. Compound 3, resulting from the addition of a phenyl ring to 2, showed further improvements in binding affinity and enzyme inhibition. Figure 2 shows a representative binding isotherm obtained by ITC for the interaction of 3 with renin.
|
Non-linear least-squares analysis of this data as described in Materials and methods, resulted in the following thermodynamic parameters: stoichiometry of binding, N= 0.86; Ka = 2.1x107 M-1 or Kd = 48 nM, and ΔH = -9.44 kcal/M. An average Kd of 79 nM for multiple binding experiments was measured by ITC that agreed closely with the renin IC50 of 58 nM. Binding enthalpy was only slightly improved compared to 1 but there was 1.8 kcal/M less entropic penalty for the binding interaction. Figure 3 shows the co-structure of 3 bound to the renin active site as determined by X-ray crystallography at a resolution of 2.6 Å. Extension of the methoxypropyl ether into the S3sp was evident.
|
|
H-bonding to the diaminopyrimidine ring was similar to 1 and the S2 pocket was unoccupied. Binding thermodynamics for several other molecules with extensions into the S3sp were examined and the results are listed in Table 1. All compounds with S3sp extensions bound tighter and inhibited renin greater than 1 but none of the compounds tested bound significantly tighter by ITC than 3. Substitution of an N-ethyl acetamide for the methoxypropyl, compound 6, resulted in a small increase in renin inhibition and binding affinity. For comparison, the thermodynamics for 5 can be examined since it differed from 6 by just a methyl substituent and the S3sp substituent. For all the compounds analyzed by ITC and listed in Table 1, there was good agreement of the renin binding affinities determined by ITC and renin inhibition, except for 7 where renin IC50 = 7.1 nM and Kd = 186 nM. Initial ideas for extending structure into the S2 pocket were provided by NMR binding experiments.25 Several small compounds were identified that bound renin in the presence of 2. One of the compounds identified was a methoxyaminophenylbenzamide.
Additional NMR experiments indicated the small molecule bound in the S2 pocket in the presence of 2 and was positioned close enough to be synthetically tethered. Compound 9 was synthesized based on this data.25 Compared to the similar compound 5 with a renin IC50 of 132 nM, 9 was less potent with an IC50 of 336 nM. This was contrary to what was initially expected since the substituent should occupy a significant portion of the hydrophobic S2 pocket. But, thermodynamics data for the binding interaction of 9 with renin helped to explain the loss of activity. Binding affinity of 9 was 272 nM compared to the tighter binding affinity of 178 nM for the similar compound 5. Interestingly, there was also a substantial loss in binding enthalpy for 9, only –2.1 kcal/M, compared to –9.3 kcal/M for 5. Based just on the reduced binding enthalpy it would have been expected that binding affinity would have dropped more substantially, but the loss was largely offset by favorable binding entropy of 6.9 kcal/M for 9.
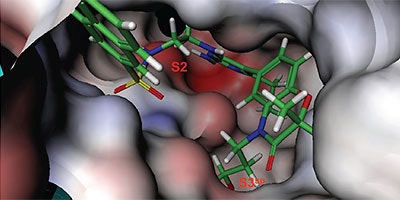
Analysis of the thermodynamics data for 9 suggested that the polar substituents that were buried in the S2 pocket were not positioned to form H-bonds and therefore significant losses in binding enthalpy occurred due to desolvation of the polar groups that were not recovered upon binding. Thermodynamics data suggested that improved affinity could be obtained by incorporation of substituents in the S2 pocket that correctly position polar functionalities to interact with the negatively and positively charged areas in the S2 pocket. Compounds 10 and 11 resulted from these initial synthetic efforts and the thermodynamics data are listed in Table 1. Compound 11 with a napthylsulfonamide substituent in the S2 pocket resulted in improved affinity, Kd = 79 nM, and renin inhibition, IC50 = 27 nM. Figure 4 shows the co-structure of 11 bound to the renin active site obtained by X-ray crystallography at a resolution of 2.24 Å.
|
Positioned in the back of the binding pocket, the diaminopyrimidine is positioned similar to previous analogs making H-bonds to Asp215 and Asp32 with the methoxypropyl substituent extending into S3sp. The napthylsulfonamide extends into the large hydrophobic S2 pocket positioning the oxygen atoms of the sulfonamide close to the positive charge at the bottom of the S2 pocket. Two different orientations of the napthylsulfonamide ring were detected in the crystal structure. In one binding orientation (Figure 4B), the sulfonamide oxygen atoms form H-bonds with the amide backbone of Ser219 and the hydroxyl side chain of Thr77. A peptide flap (renin residues Lys238–Tyr244) near the binding site was also in a closed conformation.
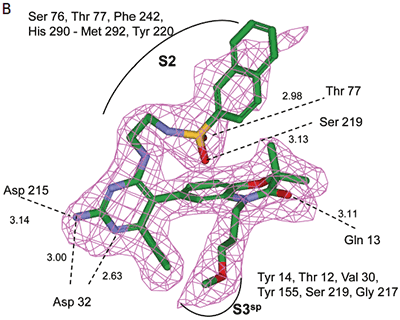
|
|
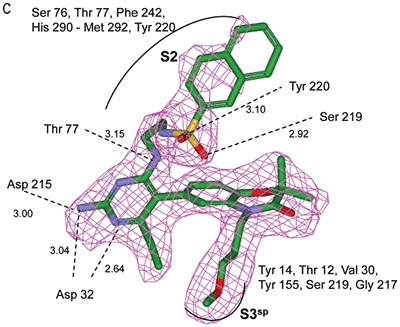
In the other ligand orientation (Figure 4C), the sulfonamide oxygen atoms form H-bonds with the amide backbone of Ser219 and Tyr220. His 290 flipped its orientation to accommodate the napthyl ring of 11, and the binding site flap was in an open conformation with a 4.2 Å shift in the position of Phe242. The electron density was clear for all the inhibitor atoms in both orientations, except the electron density of the outermost ring of the napthyl group was not well refined.
High-throughput screening of renin uncovered a substituted diaminopyrimidine that inhibited renin activity. Initial synthetic modifications produced 1 with a renin IC 50 of 6.6 μM and a K d of 3.6 μM as determined by ITC. Several hydrogen bonds were formed between the diaminopyrimidine and renin including H-bonds from the 2-amino group to Thr77 and Ser76, an H-bond between Asp215 and the 4-amino group, an H-bond between Asp32 and the 4-amino group, and an H-bond between Asp32 and the pyrimidine ring nitrogen at position 5. In addition, the binding pocket is negatively charged in the area surrounding the pyrimidine ring system and complements the basic nitrogen ring system of the inhibitor. There is a large hydrophobic pocket, S2, and a smaller hydrophobic pocket, S3 sp , that are adjacent to the catalytic site as shown in Figure 1.
Molecular modeling indicated that a methoxypropyl substituent could be tethered to the lead template to position the substituent in the S3sp. The addition of this substituent, resulting in 2, improved renin affinity to 530 nM and IC50 to 690 nM. Compound 2 was also selective versus two other aspartyl proteases, cathepsin and pepsin. Renin has low active site homology to other human aspartyl proteases. This low homology and lack of cathepsin and pepsin inhibition suggested that side effects from inhibition of other aspartyl proteases would be less likely but still needs to be addressed with in-vivo studies. The S3sp extension of 2 altered the binding thermodynamics. Extension of the methoxypropyl group into the hydrophobic S3sp was expected to expel ordered solvent molecules from the pocket resulting in favorable binding entropy. But, it was also expected that entropic contributions from the solvent exclusion would be partially offset by restricting the four rotatable bonds of the methoxypropyl ether group. Regarding the enthalpic contribution to binding free energy for the S3sp extension, the formation of favorable hydrophobic van der Waals (VDW) interactions with the ligand was expected to increase binding enthalpy. Actual thermodynamics for 2 indicated that restricting the rotatable bonds outweighed the entropic gain from solvent expulsion, but the enthalpic contribution from hydrophobic interactions outweighed that entropic penalty resulting in a net overall gain from the extension into S3sp. Compound 2 had a 5 kcal/M increase in binding enthalpy relative to 1 but this gain was offset by a 3.9 kcal/M loss in binding entropy for a net overall gain of 1 kcal/M.
Binding thermodynamics were also examined by ITC for a compound with a different substituent in S3sp. The N-ethyl formamide substituent (compound 6) extending into S3sp provide a small gain in affinity. Compared to 5, the IC50 also improved twofold. But, additional gains in affinity could be obtained for other groups extending into the S3 pocket if functionality could be added to the end of the hydrophobic extension that could interact with Tyr155 at the bottom of the S3 pocket. Compound 4 with a tricyclic ring off the diaminopyrimidine bound renin with similar thermodynamics as 5.
Compound 3 had one of the best renin affinities of the inhibitors tested. Relative to compound 2, it had a ~10 fold gain in renin inhibition and ~7 fold gain in affinity. The change in binding enthalpy for the interaction of 3 with renin was –10 kcal/M, which was 4.5 kcal/M less than for 2, but TΔS was –0.2 kcal/M compared to –5.9 kcal/M. Improved binding entropy is likely related to displacement of additional ordered solvent molecules from the binding pocket by the phenyl substituent making VDW contact with the protein.
Compound 7 with a 3,5-difluorophenyl substituent had the best IC50 of the compounds tested by ITC. Affinity determined by ITC was weaker than expected based on IC50 but 7 was tightly bound with a Kd of 186 nM. Compound 7 was bound tighter and inhibited renin better than 8 although the only difference was the methyl substituent. Structural data indicated that the 3,5-difluoro phenyl ring of 7 was positioned in closer contact with the hydrophobic surface of the binding pocket than 8 where the 3,5-difluorophenyl ring was more solvent exposed. The 3,5-difluorophenyl group of 7 was positioned to make VDW contact with Pro111. Therefore, there was increased binding enthalpy from VDW interactions. In addition, the entropic cost of restraining the phenyl group in a better binding orientation was paid at synthesis so there was less entropic penalty upon binding.
Perhaps the most interesting substituent effect was the significant loss of binding enthalpy and increase in binding entropy for compounds 9 and 10 that had extensions into the S2 pocket. Increased binding entropy is likely due to displacement of ordered solvent molecules from the large hydrophobic S2 pocket that occurs upon burial of non polar surface area for the inhibitor. Loss of binding enthalpy could be explained by burial of polar functional groups into the S2 pocket that did not form productive interactions or H-bonds. Unlike compounds 9 and 10, compound 11 with an S2 extension did not lose as much binding enthalpy but also did not add as much favorable binding entropy. The sulfonamide oxygen atoms on the napthylsulfonamide group form a polar interaction with the S2 pocket and therefore binding of 11 does not pay the same enthalpic penalty as 9 and 10. However, the added polar interaction removes some of the flexibility of the inhibitor and protein backbone resulting in less favorable binding entropy. In addition, nonoptimal binding interactions of the napthylsulfonamide group of compound 11 in the S2 pocket apparently contributed to the alternative binding modes of this group as detected in the crystal structure. In one inhibitor binding mode, a peptide flap (residues Lys238–Tyr244) near the renin active site is in a closed conformation, in the other inhibitor binding orientation, the flap is in an open conformation. The mixed population of open and closed conformations may negate a portion of entropic gain due to expulsion of ordered water molecules from the closed loop conformation. Entropy / enthalpy compensation such as seen in compounds 9, 10 and 11 can be difficult to balance in optimizing functionality for binding interactions in the S2 pocket. Results from 11 suggest that further optimization of substituents to form better positioned polar interactions in the S2 pocket could enhance binding and therefore enzyme inhibition.
The current work has shown that a combination of library screening, molecular modeling, analysis of structural, thermodynamic, and inhibition data provided a valuable tool for structure based drug design. Even when structural data are not available, thermodynamic data can provide insight into drug design but interpretation of thermodynamic data is more insightful when structural data are available. Some insight into drug design that thermodynamic data provide is intuitive to skilled medicinal chemists, but there are situations that are not apparent without thermodynamic measurements. This work used a combination of structural, thermodynamic, and inhibition data to design unique potent small molecule inhibitors of renin.
The authors would like to thank John Bryant for collecting IC50 data.
