The stability of a biopharmaceutical protein is critical to the success or failure of the development of a viable drug. Protein stability is important for production, manufacturing, formulation, long term storage, delivery to patient, and efficacy. Highly stable proteins will likely have fewer issues during the manufacturing process, are more cost-effective to produce, and are more likely to remain functional during formulation and storage without chemical alteration or aggregation. In the 'Quality by Design' (QbD) approach for biopharmaceutical development, stability characterization is part of the assessment of the 'developability' and 'drugability' of any candidate molecule, as well as during process development and manufacturing. Stability data is also incorporated in higher order structure (HOS) characterization and “fingerprinting” used for manufacturing support, biopharmaceutical comparability and biosimilarity assessment. In addition, protein HOS characterization is becoming 'expected' in regulatory submissions for new biopharmaceutical drugs and biosimilars.
Due to the complex nature of proteins, biophysical tools are important for the complete characterization of a biopharmaceutical product. There are several biophysical tools used to assess protein stability, including (but not limited to) circular dichroism (CD), dynamic and static light scattering (DLS and SLS), size-exclusion chromatography–multi-angle light scattering (SEC-MALS), Fourier transform infrared spectroscopy (FTIR), analytical ultrafiltration (AUC), size exclusion chromatography (SEC), differential scanning fluorescence (DSF), intrinsic fluorescence (IF) and differential scanning calorimetry (DSC).
While each of these biophysical assays plays an important role in biopharmaceutical development, characterizing thermal stability by DSC is critical. In a 2015 article about biophysical techniques for monoclonal antibody higher order structure characterization, Gokarn et al. stated: "DSC remains as an unparalleled technique to assess the thermodynamic stability of proteins in a given buffer condition”[1].
The intention of this whitepaper is to explain how DSC is used to characterize the thermal stability of protein biopharmaceuticals (primarily antibodies) during the selection of drug candidates to advance into development.
DSC is a microcalorimetry technique that is used to characterize the thermal and conformational stability of proteins, nucleic acids, lipids, and other biopolymers [2-7]. DSC measures heat capacity as a function of temperature. The DSC instruments used for protein characterization which are described in this whitepaper are 'power compensation' instruments, with the biopolymer in solution placed in a fixed sample cell, and corresponding buffer placed inside a matched reference cell. The heat capacity (Cp) signal from the sample cell is compared with that of the reference cell. As the temperature of the cells is increased, the temperature differences between the reference and sample cells are continuously measured and calibrated to power units. DSC is a 'forced degradation' assay: as the protein is exposed to increasing temperature, the protein begins to unfold, and the Cp of the protein increases (Figure 1).
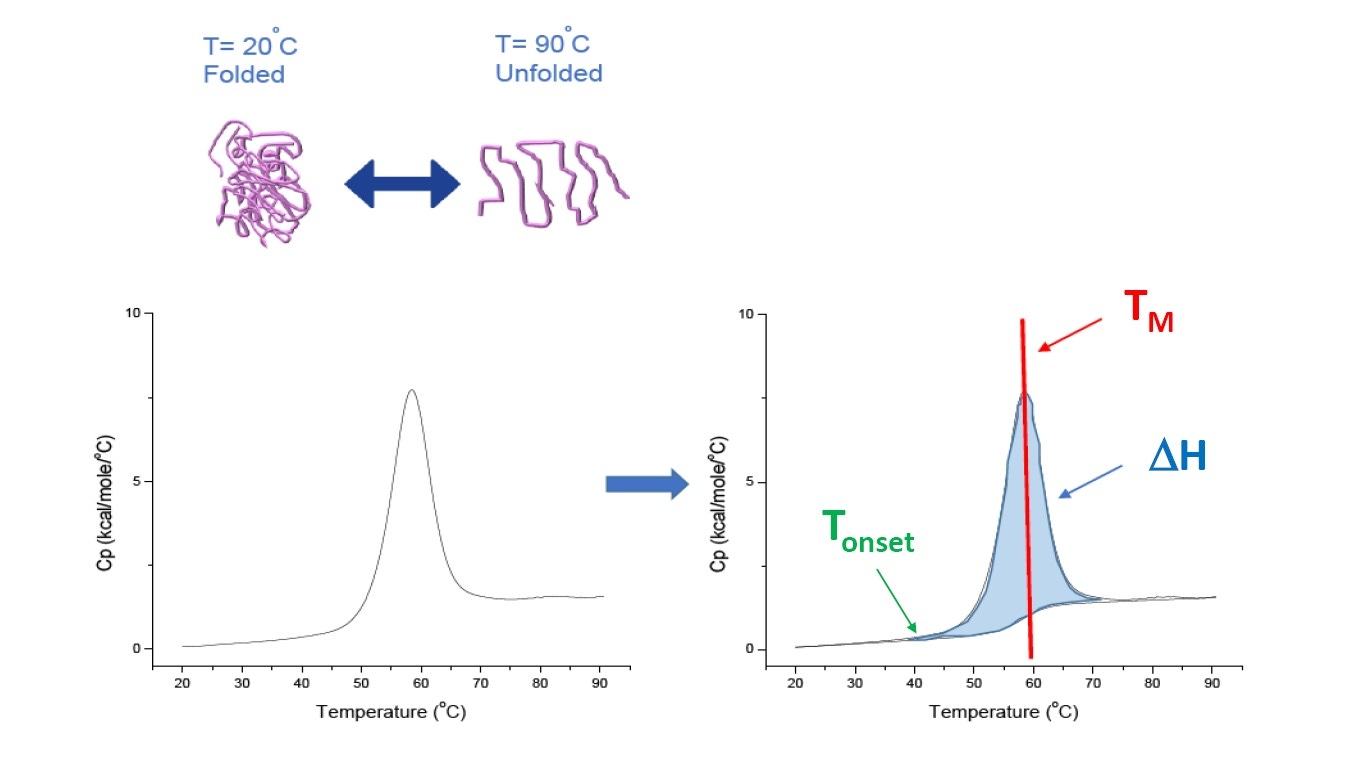
Figure 1: How DSC works.The heat capacity (Cp) changes as a protein thermally denatures. The DSC experiment starts at a temperature at which the protein is primarily folded in its native conformation. With increasing temperature, at some point the protein will begin to unfold/denature (Tonset) and the Cp increases. At the temperature at which 50% of the protein is in its native conformation, and 50% is denatured, the Cp will reach its maximum value - this is the thermal transition midpoint or TM. Above the TM, the protein will primarily be denatured, and at the end of the DSC experiment all of the protein will be in its unfolded conformation. Experimental parameters for DSC include Tonset, TM, and the unfolding enthalpy (∆H).
DSC directly measures the heat capacity change, without need for any fluorescence or other labels or probes. The thermal transition midpoint (TM), also called the melting or denaturation temperature, is the temperature at which 50% of the protein is in its native (folded) conformation, and 50% is in its denatured conformation. The TM is seen as the “peak” of a DSC thermogram (Figure 1).
TM is considered a good indicator of thermal stability – the higher the TM, the more thermally stable the protein is. Multi-domain proteins (like antibodies) typically have more than one peak on a DSC thermogram, so more than one TM can be determined (see Figure 2 for an example).
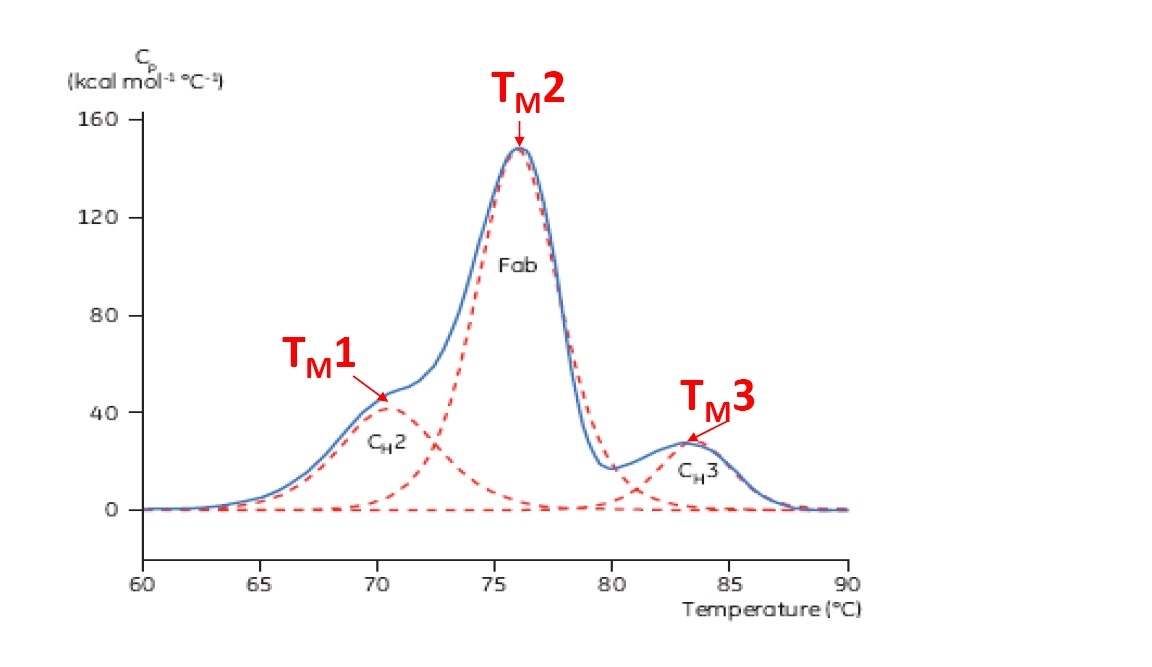
Figure 2: Representative DSC thermogram of a monoclonal antibody, with CH2, Fab, and CH3 domains identified. The dashed red lines are the deconvoluted peaks of each domain transition, with the three TMs indicated.
DSC provides other useful parameters which can be used to characterize and rank order protein stability, including the unfolding enthalpy (∆H) which is measured by the area under the curve. Protein unfolding is endothermic, since energy input is needed to break the secondary non-covalent bonds that keep the protein folded. DSC also determines the Tonset (start of unfolding), ∆Cp (heat capacity change of unfolding), and T1/2 (width at 1/2 the peak height, indicative of the shape of the unfolding thermogram). DSC analysis can include the determination of any combination of these parameters.
Malvern Instruments offers the MicroCal VP-Capillary DSC system [8,9], an automated DSC designed for TM screening and thermodynamic characterization of proteins and biopolymers in solution.
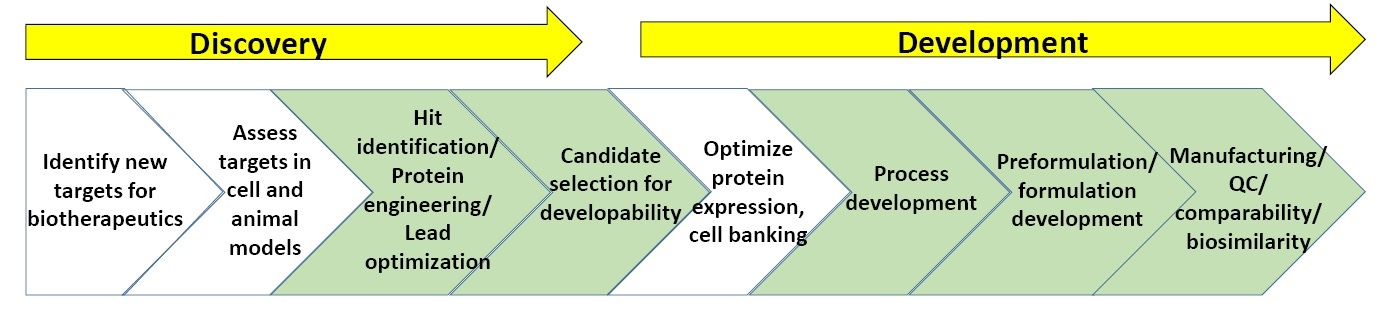
Figure 3: General scheme of processes in biopharmaceutical discovery and development.
Figure 3 shows a general scheme of the characterization processes involved in biopharmaceutical discovery and development. The sections in green show where biophysical characterization, including stability assays, is most commonly utilized. At the end of this whitepaper is a list of 'Suggested Reading' on biopharmaceutical discovery and development.
The sequence of amino acids, or primary (1°) structure, is the most basic component of the polypeptide chain and protein structure. It is important to understand and characterize the protein’s three-dimensional structure, also called its higher order structure (HOS). There are three levels of protein HOS: secondary (2°), referring to the local folding patterns of a protein’s primary structure, including α-helix, β-sheet, turns, and random coils; tertiary (3°), the 3D structure of a protein which arises from its array of secondary structural elements; and quaternary (4°), which describes structures arising from the interaction of two or more identical or different polypeptide chains.
In order to create a desirable biopharmaceutical, scientists initially look for biomolecules that already demonstrate high stability during candidate selection, and they then may need to infer increased stability via protein engineering. During purification, the protein is usually removed from conditions where it is stable, correctly folded, and active, so it is important to use the correct buffers, additives, purification, and storage conditions to keep the protein as stable as possible during this process.
When protein molecules are exposed to stresses like heat, chemicals, pH changes, pressure, mixing, and high concentration, which frequently occur during biopharmaceutical production and formulation, the protein conformation can favor the denatured (unfolded) protein. Formulated subcutaneous (SC) protein drugs must be stable and unaffected at very high protein concentrations (over 100 mg/mL) within their container closure, (e.g., vial or prefilled syringe), often for several years. Proteins in solution are also susceptible to modifications such as deamidation and oxidation that may also lead to denatured, inactive proteins.
In the case of a protein biopharmaceutical, denaturation and other modifications could result in the formation of aggregates that may confer reduced efficacy/diminished functionality to the drug. More significantly, protein aggregation has been implicated in potentially fatal immunogenic responses in patients. Using stable protein drugs will lead to more cost-effective production, and more successful, safe and effective drug products.
DSC is a measure of the conformation stability of a molecule and the changes in tertiary and quaternary structure that occur when a protein is thermally denatured, as well as the effects of intrinsic and extrinsic factors on protein stability. DSC is considered the best and most thorough and quantitative analysis of thermal stability used in the characterization of biopharmaceutical proteins, as a predictor of long-term stability[1,10-14]. TM from DSC is a parameter which is frequently used to rank-order stability in candidate selection (developability), formulation screening, and process development. More stable proteins have a higher TM. Enthalpy (∆H), Tonset, T1/2, and ∆Cp from DSC are also used in rank-ordering stability, validation of DSC data, quantitative analysis of protein unfolding, and higher order structure 'fingerprinting'[10-14].
DSC analysis of proteins in defined solution conditions is reproducible and quantitative if the proteins analyzed are the same or highly similar (Figure 4). That is, the DSC thermograms will have a reproducible pattern, and parameters (including TM, ∆H and Tonset) will be within an accepted range[12-14]. If the thermograms are different and the DSC fit parameters change, this suggests there is protein misfolding, degradation, aggregation, differences in solvent, changes in post-translational modification, or other higher order structure differences which are causing changes in conformational stability.
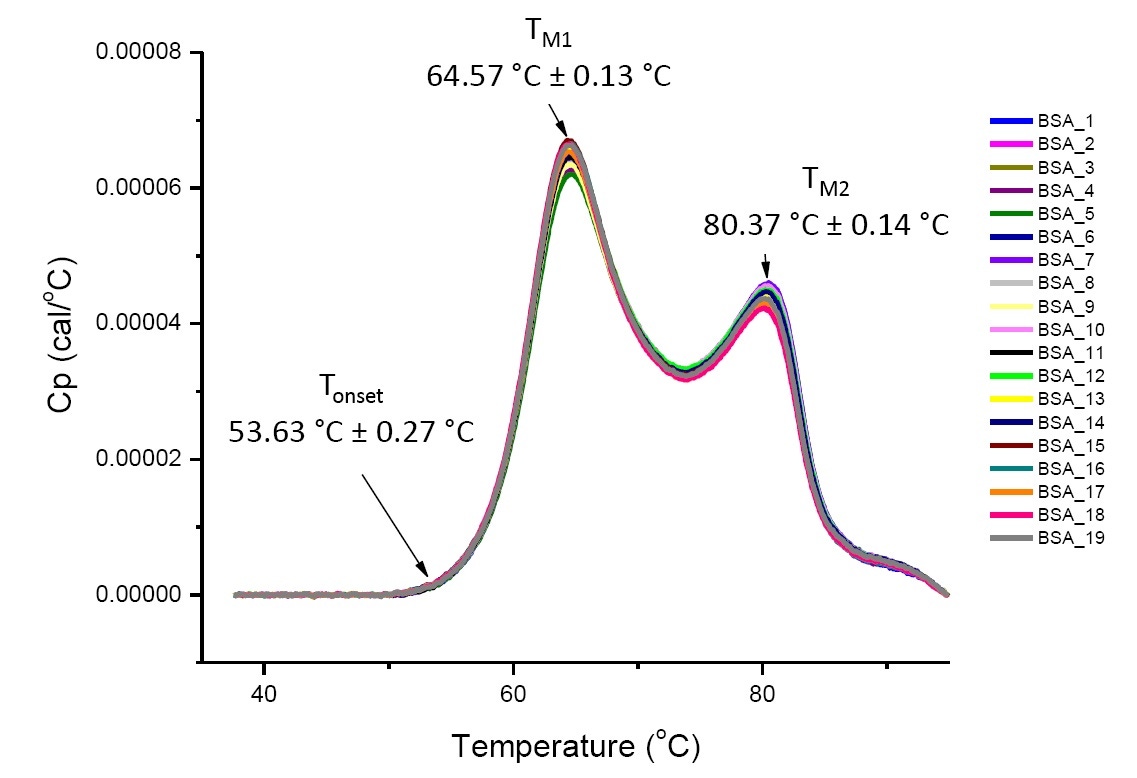
Figure 4. Nineteen DSC thermograms of bovine serum albumin (Sigma A1933, chromatographically purified) in PBS. DSC data shown after scan rate normalization, buffer-buffer subtraction, and integration baseline subtraction. Mean and standard deviations of Tonset, TM1, and TM2 are shown.
Reproducible and quantitative data makes DSC a valuable tool in HOS analysis for product evaluation during manufacturing (including batch-to-batch and site-to-site comparability), comparison of protein variants and modified products (including structural changes due to glycosylation and oxidation), and biosimilarity assessment. DSC data are also used in regulatory support documents as a HOS characterization for new drug and biosimilar submissions. In a survey of scientists in the biopharmaceutical industry, DSC was ranked as a "very useful" to "extremely useful" HOS biophysical tool for candidate selection, formulation development, product characterization, comparability, and biosimilarity[15].
For multi-domain proteins like antibodies, DSC thermograms have more than one unfolding transition (Figure 2). DSC is able to characterize and quantitate the different domains and determine the TMs for two, three or more transitions. The TM values are taken from the thermogram peak(s) and can be simply determined from the DSC data, without the need for complex data analysis. Other biophysical assays which can determine TM, such as CD, IF and DSF, may only detect the first TM (occuring at the lowest temperature), or the most "dominant" TM for multi-domain proteins. Extraction of more than one TM from spectroscopic and fluorescence data requires complex data fitting and may not be reproducible.
DSC does require more protein sample per scan, and can be lower-throughput, compared to other TM screening assays. If sample is limited, one option would be to perform an initial TM rank ordering with DSF or IF, then choose several samples to validate TM using DSC. It is important to validate the TM results with DSC, and not rely only on fluorescence or spectroscopy for TM in stability assays. Fluorescence-based assays can have artifacts which interfere with the output, and the TM results could possibly be shifted to a higher (or lower) value due to these artifacts. Some proteins and buffer conditions are not compatible with fluorescence, and TM differences may not be detected by these methods. Finally, fluorescence and spectroscopy do not determine the calorimetric enthalpy, and other thermodynamic parameters, in contrast to DSC.
DSC is considered by the biopharmaceutical industry as the “gold standard” thermal stability assay, because DSC:
Examples of how DSC data are used in biopharmaceutical discovery and development will be found in this whitepaper, other Malvern whitepapers and application notes, and cited publications.
In the research phase of identifying a protein biopharmaceutical, the initial focus is on target discovery for potential drug interaction. Once a potential target is identified, work begins on finding a drug that will interact with this target, which will elicit a desired biological effect that could lead to a favorable clinical outcome of reducing or eliminating a disease state. After one or more drug candidates have been identified, it is important to assess the “developability” of these possible biopharmaceutical candidates.
The developability of a protein drug is and evaluation of the protein’s biophysical properties to predict which protein(s) will be able to withstand the stresses they will likely encounter during cell culture production, purification, formulation, packaging, shipping, and long-term storage (stability). It is important to understand:
Performing the developability analysis of a protein drug candidate at the end of 'discovery' before preclinical development begins is important, as once the lead candidate is selected and work begins on scaling up production, any attempt to change the drug or the process will incur increased costs and increase the likelihood of the project being dropped. Making good choices at this early point results in less failures, and a significant cost reduction for the drug company as well as the production of cost-effective drugs.
At this point in late discovery/early development, there is often a limited amount of the purified protein available for the biophysical assays, and a limited amount of time for the developability assay evaluation. Biophysical methods such as fluorescence, light scattering, size-exclusion chromatography and DSC are all commonly used at this point. Other assays like analytical ultracentrifugation and hydrogen-deuterium exchange with mass spectrometry (HDX-MS) can also provide useful information.
Simple early profiling of protein drugs using DSC to rank-order TM values can be very useful in comparing drug candidates to help find proteins which have an optimal stability, to predict long-term stability, and to avoid those molecules that have less thermally stable structures. Proteins with the lowest TM values are considered the least desirable to progress as drug candidates, due to their greater instability as a function of temperature.
Doyle et al.[16] published a case study using biophysical methods to characterize bispecific Adnectin candidates during discovery, including candidate selection. Biophysical assays included: SEC, AUC, SEC-MALS, X-ray crystallography, SPR, DSF and DSC. Comparison of two Adnectin candidates by DSC showed that Candidate B showed a higher TM, and a higher Tonset (both indicating a greater thermal stability) compared to Candidate A. Candidate B also showed evidence of folding reversibility with DSC; that is, the protein sample was cooled and reheated in the DSC, and the 2ndheating cycle exhibited the same DSC profile. This reversibility also suggests improved expression levels and reduced tendency for aggregate formation.
The largest protein class of biopharmaceuticals on the market/in clinical trials is monoclonal antibodies. Monoclonal antibodies typically show a complex, multi-domain thermogram in DSC (see Figure 2 for an example). The largest, most prominent domain peak is the Fab (antigen binding fragment) of the antibody. The CH2 and/or CH3 domains are also commonly seen. The relative positions and TMs of the different domains depend on the specific monoclonal antibody, and can vary depending on subclass and engineering, as demonstrated in the below examples.
In 2007, Garber and Demarest published an article describing the use of DSC to characterize seventeen full-length therapeutic antibodies[17,18]. Using MicroCal VP-Capillary DSC, the Fabs of these antibodies exhibited thermal unfolding transitions with midpoints (TMs) varying from 57°C to 82°C. The authors demonstrated that IgG1 was the most stable human subclass, followed by IgG2 and IgG4, and for the examples used, they showed that stability variations between antibodies of a similar subclass were derived from the variable domains. The unique features of each V-gene led to a broad range of Fab stabilities.
Some antibody Fabs may be exquisitely stable while others may be relatively unstable. Very low Fab stability from TM, as observed for several constructs included in Reference 17, was related to poor/reduced antibody expression and increased aggregation issues when compared to the antibodies with increased Fab TM. As a result, it is more difficult to purify a protein with lower TM, resulting in reduced yield during process development, as well as an increased likelihood of aggregation during storage.The data from this study[17] provided evidence that DSC stability data is a useful tool in candidate selection for biopharmaceutical drugs.
Ionescu et al.[19] generated DSC profiles (using MicroCal VP-Capillary DSC) of three humanized IgG1 monoclonal antibodies and their Fab and Fc fragments (after enzymatic digestion) at neutral pH. With some exceptions, the thermograms showed that the transition with the larger experimental enthalpy contained the contribution from the Fab fragments (similar to the results in reference 17). Also, the apparent TMs were found to vary significantly, even for Fab fragments originating from the same human germline. Ionescu at al. proposed using the unfolding enthalpy as the key parameter to recognize the unfolding events in the melting profile of an intact IgG1 antibody. Some DSC thermograms showed two transitions: the first transition representing the unfolding of the Fab fragment and the CH2 domain, and the second transition representing CH3 domain unfolding. In other antibodies, the first DSC transition represents CH2 domain unfolding, and the second transition represents the unfolding of the Fab fragment and the CH3 domain. There were other cases where the DSC profile presented three transitions, with the Fab unfolding occurring at distinct temperatures compared to the melting of the CH2 and CH3 domains. Low stability or heterogeneity of the Fab fragment may prove problematic for long-term storage or consistency of production. The authors proposed that fully understanding the features of a DSC profile is important for candidate selection in the early stages of development of therapeutic monoclonal antibodies.
The work described by the two papers above, as well as others published during the same time period, formed the groundwork for subsequent case studies and the use of DSC and other stability characterization studies as part of biopharmaceutical candidate selection.
To further delineate the thermal stability of drug candidates, proteins are frequently analyzed at more than one pH condition to see if any differences arise. These different pHs can represent conditions encountered in formulation and/or process buffers used during the manufacturing of the drug.
Jiang et al.[20] published a case study using selected HOS characterization to evaluate the developability (ability to manufacture, as well as maintain overall product quality) of two potential monoclonal antibody drugs. Antibodies X and Y were IgG2 mAbs against the same target, which exhibited similar biological activities. Using a combination of biophysical techniques (including DSC, near UV CD, FTIR, DLS, fluorescence spectroscopy, and SEC): they looked at:
The acidic condition (pH 3) was chosen to replicate buffers used during antibody purification and viral inactivation, the pH 5 buffer simulated storage conditions (formulation) and pH 7 buffers were used throughout purification.
MicroCal VP-Capillary DSC scans of the two mAb candidates in pH 7 buffer (PBS) showed two transitions: the CH2/Fab domains represented the 1st transition, while the CH3 domain unfolding was represented by the 2nd transition[20]. mAb Y was slightly more thermally stable at pH 7, based on higher TMs from DSC as well as higher onset temperature (Tonset). In pH 3 buffer (C3N), there was only one DSC thermal transition for each candidate, suggesting that the samples were already partially unfolded at the starting temperature, due to the low pH. The thermal transition temperatures and enthalpies of unfolding of both candidates in pH 3 were markedly lower than those in PBS, as expected. However, mAb X (TM of 63.2°C) showed better thermal stability than mAb Y (TM of 48.2°C) in C3N, even though mAb Y was more thermally stable at neutral pH. Although this result is atypical, it is consistent with the near UV CD data, which showed that mAb X retained more tertiary structure at pH 3. Changes in the thermal stability of the two candidates induced by pH 3 treatment appeared to be fully reversible following dialysis into PBS.
Overall, the DSC results indicated that both proteins were very stable at neutral pH, and the thermal stability of mAb Y was higher than that of mAb X in PBS, but lower than that of mAbX in C3N[20]. CD and DSC data showed that mAb Y underwent more structural changes at pH 3 than did mAb X. Thermal stability changes induced at pH 3 were fully reversible for both mAbs, although tertiary structural changes appeared to be at least partially irreversible for mAb Y. Compared with mAb X, mAb Y showed better thermal stability at neutral pH and improved storage stability at 37°C in its formulation buffer at pH 5. Taken together, the results suggest that there are different driving forces for process stability (robustness to pH changes) and storage stability (impact of elevated temperature).
Because folded mAb Y had higher thermal stability in PBS at pH 7 and better stability at pH 5 during storage at 37°C, mAb Y was recommended to be progressed for further development[20]. Also, due to the irreversibility of both tertiary structure disruption and then self-association in PBS that was observed after low pH incubation, the recommendation to advance mAb Y was accompanied by a recommendation to develop the purification process to minimize exposure of mAb Y to pH 3. This case study demonstrated how stability data as part of candidate selection and process development helps minimize the amount of irreversible aggregate and conformational variant produced by the process, and will ultimately increase the process yield[20].
Several more case studies using DSC thermal stability to evaluate and predict developability are summarized in Satish et al.[21]
Tavakoli-Kenshe et al.[22] wanted to see which other predictors besides stability studies could be used to predict developability. They looked at five variants of IgG1 and IgG4 antibodies, and used a rotating disk shear device which produced defined shear conditions at a known solid–liquid interface to measure stability in this environment. Antibodies were ranked for stability based on shear device output and compared with accelerated thermal stability data and the melting temperature of the CH2 domain (TM1) from MicroCal VP-Capillary DSC to investigate technique complementarity. Results suggested that the techniques are orthogonal, with thermal methods based on intramolecular interaction, and shear device stability based on localized unfolding, revealing less stable regions that drive aggregation. Molecular modeling shows the effects of the modifications on the antibody structures and indicates a possible role for Fc conformation and Fab-Fc docking in determining suspended protein stability. The data introduces shear testing as a potential orthogonal stability indicator, complementary to traditional thermal methods like DSC, allowing candidate selection of proteins with improved stability for the purification process.
Frequently in biopharmaceutical discovery, the 'hit' molecules or 'parental' protein drug molecules are modified or engineered to improve their biophysical characteristics and make the protein more developable. After engineering, prior to candidate selection, it is important to evaluate the different engineered proteins for stability, to see if engineering resulted in any deleterious effects. One example of this was presented in a Malvern application note[23], where MicroCal VP-Capillary DSC was used as a screening tool for the parent antibody and two engineered antibodies, to help predict the thermal stability and developability of the engineered constructs. Aggregation formation (via SEC) after accelerated storage was also studied. The engineered antibody with the most-decreased TM correlated to the antibody construct with the highest amount of aggregation after accelerated stability screening, assisting the guidance of candidate selection.
Demarest et al. engineered an unstable Fab and used MicroCal VP-Capillary DSC to look at improved protein stability as a demonstration[18, 24]. In an effort to develop antibody and antibody fragment stabilization strategies, they selected a poorly-behaved Fab that recognized tetanus toxin (αTT) originally isolated from a human source, and performed a mutagenesis campaign to engineer stability back into the Fab. The authors chose 45 residue positions for randomization, and close to 4500 individual colonies were picked and cultured in expression media. Supernatants containing expressed Fab were heat-challenged at three elevated temperatures (70°C, 72°C, and 74°C). Variants that demonstrated enhanced thermostability were resubjected in duplicate to the thermotolerance screen to confirm their properties (see reference 24 for complete list of stabilizing mutants). Approximately 1% of the variants in the library exhibited enhanced thermostability. Fourteen of the 'hits' were located in the VH domain, and the remaining four in the VL domain. This result suggested that the stability of the native Fab was limited by the marginal stability of the VH. Surprisingly, no mutant within the ~2000-member constant domain library was found to stabilize the Fab as a whole. The authors speculated that stabilizing mutations within the constant domains really do occur, but that the limited stability of the VH domain controls the temperature at which the Fab unfolds, and limited the ability to observe such events.
Four stabilizing mutants were discovered within the αTT VL domain[24]. Mutation of the VK4 consensus residue W50 to Ala (the consensus residue for VK1) or His was highly stabilizing. Histidine is rarely found at position 50 in human kappa variable domains, but is often found in human lambda variable domains. This residue is close to the VH/VL domain interface. The authors hypothesized that its contribution to Fab stability might be linked to buttressing of the VH domain, since the VH domain in particular appears to limit the stability of the αTT Fab. However, studies with the isolated VL domain suggested otherwise.
Twelve constructs were generated containing between three and eleven stabilizing mutations which were identified in the initial screen[24]. Various combinations were derived rationally to determine the apparent contribution each mutant provided to Fab stabilization. Introduction of multiple stabilizing mutations to the αTT Fab increased Fab expression in a parallel transformation/expression experiment, suggesting the more stable constructs resulted in increased expression in the cells. The best constructs consistently exhibited more than three-fold increases in expressed yield over wild-type. The stability of each Fab was evaluated by DSC and circular dichroism (CD). The measured TM values were used to rank-order the apparent Fab stability.
One important consideration was the potential effect these mutations might have on the functional antigen binding of the αTT Fab. The thermostabilizing mutations were derived from the original screen using a quantitative ELISA, detecting both the CL domain and the histidine tag at the C terminus of CH1. The authors saw that the thermostabilizing mutations enhanced the apparent affinity of the αTT Fab in a functional ELISA, compared to wild-type[24]. The functional capacity of each Fab variant appeared to be directly correlated to its stability as described by the TM from DSC. The ability to correlate functional activity and protein expression to the TM from DSC suggested that choosing the most stable construct will lead to improved developability.
Seeliger et al.[25] incorporated computational design strategies to systematically modify an antibody that exhibited a tendency to aggregate in vitro. The resulting series of closely related antibodies engineered from a wild-type (WT) mAb1 showed improved stability as assessed by biophysical methods (including MicroCal VP-Capillary DSC) as well as long-term stability experiments. As Demarest et al. noted in the above example with αTT Fab[24], mutant protein expression levels also improved in comparison with the wild-type candidate. Together, the experimental and computational data used in this study demonstrates how computational methods can be used to guide antibody optimization for increased stability.
DSC of the original wild-type protein showed a TM of 68°C for the first transition of the thermogram (identified as the Fab domain). In DSC thermograms for heavy chain or light chain mutations, the TMfor the first thermal transition (Fab) from DSC increased to 68.9°C - 72.8°C. In the mutant with engineered heavy and light chains, the first endotherm is observed at 70.5°C, with the larger peak (Fab) at 83.5°C. The TMfor the antibody Fab unfolding increases about 16 K after removal of predicted 'liabilities' in heavy chain and light chain. When unfolding was monitored via the change in tryptophan fluorescence, the same trend was observed[25].
The computer simulation predicted that most introduced mutations increase thermodynamic stability. These results were in line with those obtained from the DSC and right-angle light scattering (RALS) measurements. Both experiments demonstrated a continuous increase in stability with an increasing number of mutations. From both experiments, the authors also found that engineering the light chain had a notable effect on stability, which is in agreement with the calculated changes of the thermostability for each mutation[25].
All engineered variants of mAb1 were expressed with titers above 50 mg/L, whereas with wild-type, mAb1 titers were below 1 mg/L. The dramatic increase of the expression levels for the variants harboring only one engineered chain were not predictable from the DSC results, which showed a small stability increase for these variants. However, the calculations suggest that the stability of the individual immunoglobulin domains is improved with the introduced mutations. In experiments involving heating, it is plausible that the stability of each individual chain is the limiting factor, indicating that the observed effects were kinetic rather than equilibrium thermodynamic effects. As soon as one domain unfolds, the stabilizing effect of the complex formation vanishes, and, as a consequence, the second chain starts to unfold as well. For protein production and folding in vivo, the presence of one stably-folded domain to serve as a nucleation point for a less stable chain could be sufficient to substantially increase the amount of successfully folded antibody[25].
From the perspective of a company developing a biopharmaceutical drug, does the step-wise increased protein stability (from DSC and other biophysical assays), translate into step-wise improved long-term stability and into improved shelf-life? The authors conducted an accelerated stability study at 40 °C with the engineered variants, and they observed substantial improvements in long-term stability as judged by monomer content from SEC. The fully-engineered variant was also the most stable over the entire test period of 4 months[23]. The thermodynamic stability of the variable domains certainly is an important factor for long-term stability. But it is also evident from the authors' data, as well as other published data, that thermodynamic stability and shelf-life cannot easily be correlated in every case, and there may need to be adjustments made to improve stability in the formulation stage[25,26,27,28].
Recent publications describe rational engineering and the use of DSC and other biophysical tools to characterize protein stability, including:
The National Institute of Standards and Technology monoclonal antibody (NISTmAb) reference material[35] was evaluated for developability with HOS biophysical tools typically used for mAb drug candidates. NISTmAb is not intended to be a therapeutic molecule, and the developability assessment of NISTmAb was taken on as a class-specific, representative IgG1 mAb[36]. The NISTmAb was evaluated under different buffer conditions for stability and integrity using electrophoresis, SEC-MALS, DLS, and DSC methods. The thermal stability of the NISTmAb sample in PBS buffer was determined by MicroCal VP-Capillary DSC. The NIST sample shows three main thermal transitions assigned as the CH2 domain at 71.2°C, the CH3 domain at 84.1°C, and the Fab domain at 88.9°C.
Compared to the DSC profiles of IgG1 molecules from reference 17, the NIST Fab is more stable (88.9°C); therefore, it was concluded that the molecule has a favorable HOS profile[36]. Based upon the complete biophysical analysis data package, NISTmAb has a favorable developability profile and the changes observed in the molecule after stress conditions do not represent risks that cannot be mitigated with the right formulation and manufacturing at later stages in the development process.
Results presented in this whitepaper clearly demonstrate the importance and effectiveness of incorporating DSC as a biophysical stability assay during biopharmaceutical candidate selection. Using DSC results, along with those of other stability assays, biopharmaceutical companies can make informed decisions on the most stable and developable drug candidates, which means those with the best chances for production and purification, and those less likely to exhibit long-term stability and aggregation issues for the final formulation and drug product. This translates into more cost-effective drug production, and increased likelihood that the final drug formulation will remain active, stable, and in its correctly folded conformation.
Analytical Techniques for Biopharmaceutical Development, R. Rodriguez-Diaz, T. Wehr, S. Tuck (eds.), Taylor & Francis, New York USA (2005).
Biophysical Characterization of Proteins in Developing Biopharmaceuticals, D.J. Houde, S.A. Berkowitz (eds.), Elsevier, Amsterdam, Netherlands (2015).
Biophysical Methods for Biotherapeutics: Discovery and Development Applications, T.K. Das (ed.) John Wiley & Sons, Hoboken NJ USA (2014).
Biophysics for Therapeutic Protein Development, L.O. Nahri (ed.), Springer New York, USA (2013).
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1176 (2014). doi: 10.1021/bk-2014-1176.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1201 (2015). doi: 10.1021/bk-2015-1201.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 3. Defining the Next Generation of Analytical and Biophysical Techniques, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1202 (2015) DOI: 10.1021/bk-2015-1202.
