In this application note, triple detection size exclusion chromatography (SEC-TD) is used to understand the ability of proteins to form ordered, structured complexes. The controlling mechanism behind these protein complexes is fundamental to our knowledge of protein signaling pathways and the physiological responses they control.
The ability of proteins to form ordered, structured complexes is central to their biological function. Such complexes, which can be homo-oligomers or hetero-oligomers, can control different attributes such as activity, stability and localization. Understanding how these complexes form, and the controlling mechanism behind them, is fundamental to our knowledge of protein signaling pathways and the physiological responses they control. Of particular interest in this field, is the assembly of virulence factors complexes from pathogenic microbes. These factors are secreted by such micro-organisms to facilitate infection, survival and/or colonization of the host and are therefore crucial for the onset of disease. The ability to target and inhibit these virulence factors could lead to the development of a new generation of anti-microbial therapies. A prerequisite is to understand the mechanisms leading to the formation of hetero- and homo-oligomers. In order to characterize the protein-protein interactions of selected microbial virulence factors, advanced light scattering detectors, combined with liquid chromatography methods, were employed.
Materials and proteins were prepared as described (see References 1, 2, 3, 4).
Intrinsic viscosity and molecular mass measurements were performed by SEC-TD, with a Size Exclusion Chromatography (SEC) system connected to a triple detector array, TDA (Malvern, UK). The TDA contained (i) a static light scattering cell with two photodiode detectors, at 7° for low angle (LALS) and at 90° for right angle laser light scattering (RALS), (ii) a differential refractive index detector, (iii) a photometer, and (iv) a differential viscometer. Protein concentration was determined using both the photometer and the refractive index detector. The RALS and LALS data, in combination with the concentration, provided the molecular mass M. Intrinsic viscosity (η) was calculated using the differential viscometer. Both molecular mass and intrinsic viscosity were calculated with the OmniSEC software.
The conformation and formation of protein complexes of bacterial virulence factors were studied. The utilization of Size Exclusion Chromatography, coupled to a Triple Detector Array, yielded key information regarding the behavior of these proteins, as described below.
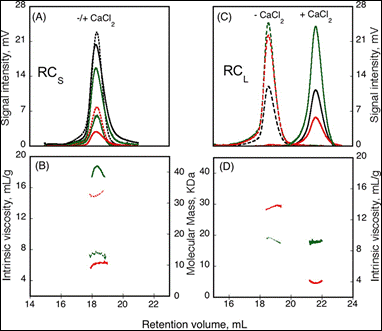
The formation of homo-oligomers can be controlled by many factors. Here, the impact of calcium binding, or the presence of regulating peptides, was studied. The “Repeat in Toxin” (RTX) motif is present in many virulence factors that are secreted by pathogenic microbes. The calcium-dependent adenylate cyclase toxin from Bordetella pertussis (CyaA) utilizes the RTX repeat domain (RD) to elicit the calcium response. Two RD subdomains, RCS and RCL (2) were characterized for their response to Ca2+ binding (Figure 1).
|
Based on the right angle scattering data (green traces) and the differential refractometer (black traces), it can be deduced that the RCL construct has the same molecular weight in both Apo (- Ca2+) and Holo (+ Ca2+) forms (Figure 1). However, the differences in retention volumes, 18 mL and 22 mL for Apo and Holo forms, respectively, demonstrate that the peptide has a smaller hydrodynamic volume when bound to calcium. In addition, the binding of calcium induces a drop in the intrinsic viscosity from 13.7 to 3.9 mL.g-1. In contrast to the RCL construct, binding of calcium to RCS did not change the retention volume, but resulted in an increase in molecular weight, from 15.1 kDa to 42 kDa, and a reduction in intrinsic viscosity from 14.7 to 6.4 mL.g-1. Taken together, these data indicate that following binding of calcium, RCL adopts a smaller, tightly formed monomeric conformation, while RCS forms a tightly structured trimer of the same size as the unfolded monomer observed in the absence of calcium.
|
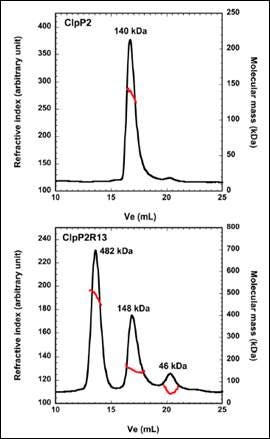
ClpP2 is an ATP-dependent proteolytic complex that is thought to contribute to the ability of Mycobacterium tuberculosis to remain dormant in host cell phagosomes for many years. In vivo, ClpP2 forms a tetradecameric complex that is required for full peptidase activity, although how this complex is formed and what the controlling mechanism is, remains unclear. A disordered N-terminal peptide sequence had been proposed to play a role in regulation. Therefore, wild type ClpP2 and an N-terminal truncated form, ClpP2R13, were analyzed using size exclusion chromatography with triple detection. These data are shown in Figure 2. ClpP2 eluted as a single main peak with a molecular weight of 140 kDa. This corresponds to a combination of hexamers and heptamers. However, removal of the N-terminal prosequence resulted in three main peaks at 482kDa, 148 kDa and 46 kDa. The largest molecular weight species corresponds to a 21-mer homo-oligomer. Whilst this is larger than the tetradecamer expected, these data point towards the prosequence of ClpP2 as a regulator in the formation of higher order oligomers.
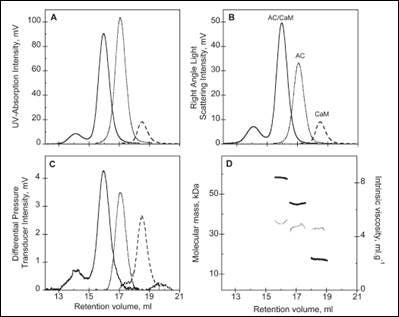
The activity of many proteins is controlled by the binding of different regulator proteins, either activators or inhibitors. Here, the complex of bacterial virulence factors with specific regulating proteins was studied with the aid of SEC-TD. CyaA is a key virulence factor for Bordetella pertussis, the causative agent of whooping cough. Following binding and activation by Calmodulin (CaM), CyaA induces the production of pathogenic levels of cAMP.
|
SEC-TD data of the individual components, and the hetero-complex, are shown in Figure 3. The molecular weight for the CaM-CyaA catalytic domain (AC) complex was determined as 58.4 kDa, which is approximately equal to the sum of the molecular weight for the monomers (43.8 kDa and 17.2 kDa for AC and CaM, respectively). These data strongly suggest the complex consists of CaM and AC at a 1:1 stoichiometry, a conclusion that is supported by Analytical UltraCentrifugation (AUC) data (not shown). Intrinsic viscosity data from SEC-TD analysis produced values of 5.2, 4.9 and 5.0 mL.g-1 for AC, CaM and CaM-AC complex, respectively. The low value for the CaM-AC complex, suggests a strong compaction of CaM and AC when the complex forms. Consistently, the hydrodynamic radius of CaM-AC, as determined by AUC, was 3.3 nm rather than the expected 3.7 nm (data not shown). Taken together, these data point towards a signaling pathway where CaM binds to CyaA, forming a compact 1:1 heterodimer.
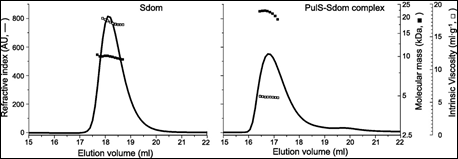
Finally. the membrane secretin of the micro-organism Klebsiella oxytoca, PulD, is a component of a multimeric membrane complex required for efficient secretion of virulence factors into the host. In order to correctly localize to the bacterial membrane, PulD associates with the chaperone protein PulS. The interaction of PulS with the S domain of PulD (Sdom) was studied using SEC-TD. Data are shown in Figure 4.
|
Molecular weight measurements confirmed the formation of a Sdom:PulS 1:1 complex, of 22.1 kDa. The intrinsic viscosity data for Sdom and the Sdom:PulS complex were 16.9 and 5.9 mL.g-1 respectively. These data, coupled to the intrinsic viscosity values of 4.4 for Sdom and 2.5 for Sdom:PulS, strongly suggest that Sdom forms an elongated disordered structure, while the Sdom:PulS complex forms a compacted, ordered structure. These data are supported by hydrodynamic radii values of 1.7 nm and 2.5 nm for PulS and Sdom respectively, while that of the Sdom:PulS complex is only 2.7 nm.
Protein signaling pathways are generally intricate, involving many different components and often multi-protein signaling complexes. Understanding the mechanisms behind the formation of hetero-oligomers and homo-oligomers is central to understanding the regulation and activity of such complexes. The data presented in this application note demonstrates the compelling information that can be obtained from the use of SEC-TD systems. Such information, including the molecular weight and size of monomers and complexes, as well as protein structure data, greatly facilitates elucidation of the mechanisms and characteristics behind these multi-protein signaling pathways.
Work contained within this application note is further described in the following publications: Biochem, (2010) Vol.49, p.318-328; J. Biol. Chem, (2011) Vol.286, p.16997-17004; J. Biol. Chem. (2011) Vol. 286, p.38833 – 38843; BMC Biochem (2011) Vol.12, Issue 61.
