このアプリケーションノートでは、ゼータサイザーナノZSP を使用したタンパク質電気泳動移動度の測定を説明しています。 拡散障壁手法と正しい測定プロトコルを使用することにより、正確に測定できるようになります。
ゼータサイザー ナノは、流体力学的直径を求めるための動的光散乱法と電気泳動移動度に対する電気泳動光散乱技術で市場をリードしています。 動的光散乱法(DLS)は、タンパク質の特性評価と製剤設計に広く採用されている手法で、分子サイズの判別に加え、安定性の評価にも使用できます。 近年では、バイオ医薬としてタンパク質の使用頻度が劇的に高まっているため、溶液内のタンパク質の安定性が科学面および商業面で大きな関心を引いています。 安全で採算の合う製品を開発するには、製剤の安定性と挙動に関する知識が欠かせないため、製剤の特性を評価できるツールの需要が高まっています。 電気泳動光散乱 (ELS) で測定されるタンパク質の電気泳動移動度は、製剤の安定性、粘度、挙動を確実に示すものとみなされてきた特性です [1]。
実験上では、タンパク質の電気泳動移動度の測定には、2 つの実質的な課題があります。 (i) 多くの場合、タンパク質溶液の処理とは、濃度希釈の操作のことです。 DLS の観点からは、低いレベルの光を比例的に分散させるサイズの小さな分子を少量扱うということです。 これまで、市場に出回っている光散乱ベースの装置の大半は、こうした測定に必要な感度を備えていません。 (ii) タンパク質の電気泳動移動度測定を行うには、試料に電界を印加する必要があります。これはそれ自体、凝集を促すことでタンパク質を破損させる可能性がある物理プロセスです [2]。 その結果、電気泳動移動度測定は、対象のタンパク質ではなく、凝集分子の電気泳動移動度を反映したものになります。
タンパク質を正確に、かつ高い品質で測定するには、次の 3 つの条件を満たす必要があります。

|
ゼータサイザー ナノ ZSP (ZSP、図 1A) と関連ソフトウェアは、これらの条件を満たすために開発された製品です。 リゾチームの場合、測定濃度の下限は1 mg/mL となり、電気泳動移動度測定における光学系の感度は、小さな径の粒子でも、低濃度試料でも解析できます。 さらに、ZSP で使用されるPALS技術(Phase Analysis Light Scattering) により、タンパク質などの低電気泳動移動度試料に対する測定性能も向上しています。
マルバーンの科学者たちの取り組みから、電気泳動測定時の凝集の多くは、電極で発生していることが明らかになっています [3]。 マルバーンが発明し、特許を取得した拡散障壁手法 (図 1B) では、セルの電極からタンパク質試料を分離して試料を保護します [2, 4]。 つまり、より信頼性の高いデータを測定で得るために、電圧をより長時間、印加できます [3]。 拡散障壁手法は、ナノサイズの生物材料の電気泳動移動度測定に関する最新の ASTM 規格で参照できます [2]。
ゼータサイザー ナノ ZSP 用のゼータサイザーソフトウェアには、専用のタンパク質電気泳動移動度測定手順も含まれています。 この測定は、電圧を抑え、セル内の温度を注意深く制御することで、試料が凝集しないように保護します。 電気泳動移動度測定の前後に、同じ光学系を使用して粒子径測定も行います (図 2)。 そのため、測定される粒度分布は、測定した電気泳動移動度の母集団を直接表します。 電気泳動移動度に対して異なる光学系で粒子径が測定された場合、光散乱強度が変化し、形成される凝集が隠される可能性があるため、これは利点となります。 ゼータサイザー ナノ ZSP は、電気泳動移動度測定の前後で同じ光学系で粒子径測定を行い、測定中に発生した可能性がある凝集を特定します。

|
最終的に、タンパク質の電気泳動移動度を正確に測定することで、タンパク質の電気泳動移動度と電荷との既知の関係に基づき、タンパク質の電荷を計算できるようになります [5]。 ゼータサイザーソフトウェアの新しい解析機能を使用すると、これらの計算を行うことができます。
本文献では、ZSP を使用したタンパク質電気泳動移動度の測定を説明します。 ZSP の感度、拡散障壁手法の適切な実施、適切な測定手順により、従来と比較して、正確な値を得ることができるようになりました。
ヒト血清アルブミン(HSA)を本来の緩衝剤(pH 7)とクエン酸緩衝剤(pH 4.3)で それぞれ約2mg/mLの濃度に調整しました。 測定の開始時点で凝集が存在しないように、0.02 µm フィルタで試料をろ過しました。
試料がセルの電極に触れることで凝集が形成されないよう拡散障壁手法を使用しました。 使い捨ての U 字キャピラリーセルに適切な緩衝剤を充填しました。 20 µLのHSAサンプルをゲルローディングチップでセル下部のレーザー透過部に添加しました。 セルを装置に挿入し粒子径測定とゼータ電位の測定を実行しました。
ゼータサイザー ナノ ZSP を使用して、HSA の流体力学的サイズと電気泳動移動度をそれぞれ、動的光散乱と電気泳動光散乱で測定しました。 測定されたゼータ電位が凝集体ではなくタンパク質であることを確認するために、粒子径と電気泳動移動度を同じ散乱角度で測定しました。
▼結果・考察は、ページ下部より会員登録の上、ログインしてご覧ください。
ゼータサイザー ナノは、流体力学的直径を求めるための動的光散乱法と電気泳動移動度に対する電気泳動光散乱技術で市場をリードしています。 動的光散乱法(DLS)は、タンパク質の特性評価と製剤設計に広く採用されている手法で、分子サイズの判別に加え、安定性の評価にも使用できます。 近年では、バイオ医薬としてタンパク質の使用頻度が劇的に高まっているため、溶液内のタンパク質の安定性が科学面および商業面で大きな関心を引いています。 安全で採算の合う製品を開発するには、製剤の安定性と挙動に関する知識が欠かせないため、製剤の特性を評価できるツールの需要が高まっています。 電気泳動光散乱 (ELS) で測定されるタンパク質の電気泳動移動度は、製剤の安定性、粘度、挙動を確実に示すものとみなされてきた特性です [1]。
実験上では、タンパク質の電気泳動移動度の測定には、2 つの実質的な課題があります。 (i) 多くの場合、タンパク質溶液の処理とは、濃度希釈の操作のことです。 DLS の観点からは、低いレベルの光を比例的に分散させるサイズの小さな分子を少量扱うということです。 これまで、市場に出回っている光散乱ベースの装置の大半は、こうした測定に必要な感度を備えていません。 (ii) タンパク質の電気泳動移動度測定を行うには、試料に電界を印加する必要があります。これはそれ自体、凝集を促すことでタンパク質を破損させる可能性がある物理プロセスです [2]。 その結果、電気泳動移動度測定は、対象のタンパク質ではなく、凝集分子の電気泳動移動度を反映したものになります。
タンパク質を正確に、かつ高い品質で測定するには、次の 3 つの条件を満たす必要があります。
|
|
ゼータサイザー ナノ ZSP (ZSP、図 1A) と関連ソフトウェアは、これらの条件を満たすために開発された製品です。 リゾチームの場合、測定濃度の下限は1 mg/mL となり、電気泳動移動度測定における光学系の感度は、小さな径の粒子でも、低濃度試料でも解析できます。 さらに、ZSP で使用されるPALS技術(Phase Analysis Light Scattering) により、タンパク質などの低電気泳動移動度試料に対する測定性能も向上しています。
マルバーンの科学者たちの取り組みから、電気泳動測定時の凝集の多くは、電極で発生していることが明らかになっています [3]。 マルバーンが発明し、特許を取得した拡散障壁手法 (図 1B) では、セルの電極からタンパク質試料を分離して試料を保護します [2, 4]。 つまり、より信頼性の高いデータを測定で得るために、電圧をより長時間、印加できます [3]。 拡散障壁手法は、ナノサイズの生物材料の電気泳動移動度測定に関する最新の ASTM 規格で参照できます [2]。
ゼータサイザー ナノ ZSP 用のゼータサイザーソフトウェアには、専用のタンパク質電気泳動移動度測定手順も含まれています。 この測定は、電圧を抑え、セル内の温度を注意深く制御することで、試料が凝集しないように保護します。 電気泳動移動度測定の前後に、同じ光学系を使用して粒子径測定も行います (図 2)。 そのため、測定される粒度分布は、測定した電気泳動移動度の母集団を直接表します。 電気泳動移動度に対して異なる光学系で粒子径が測定された場合、光散乱強度が変化し、形成される凝集が隠される可能性があるため、これは利点となります。 ゼータサイザー ナノ ZSP は、電気泳動移動度測定の前後で同じ光学系で粒子径測定を行い、測定中に発生した可能性がある凝集を特定します。
|
|
最終的に、タンパク質の電気泳動移動度を正確に測定することで、タンパク質の電気泳動移動度と電荷との既知の関係に基づき、タンパク質の電荷を計算できるようになります [5]。 ゼータサイザーソフトウェアの新しい解析機能を使用すると、これらの計算を行うことができます。
本文献では、ZSP を使用したタンパク質電気泳動移動度の測定を説明します。 ZSP の感度、拡散障壁手法の適切な実施、適切な測定手順により、従来と比較して、正確な値を得ることができるようになりました。
ヒト血清アルブミン(HSA)を本来の緩衝剤(pH 7)とクエン酸緩衝剤(pH 4.3)で それぞれ約2mg/mLの濃度に調整しました。 測定の開始時点で凝集が存在しないように、0.02 µm フィルタで試料をろ過しました。
試料がセルの電極に触れることで凝集が形成されないよう拡散障壁手法を使用しました。 使い捨ての U 字キャピラリーセルに適切な緩衝剤を充填しました。 20 µLのHSAサンプルをゲルローディングチップでセル下部のレーザー透過部に添加しました。 セルを装置に挿入し粒子径測定とゼータ電位の測定を実行しました。
ゼータサイザー ナノ ZSP を使用して、HSA の流体力学的サイズと電気泳動移動度をそれぞれ、動的光散乱と電気泳動光散乱で測定しました。 測定されたゼータ電位が凝集体ではなくタンパク質であることを確認するために、粒子径と電気泳動移動度を同じ散乱角度で測定しました。
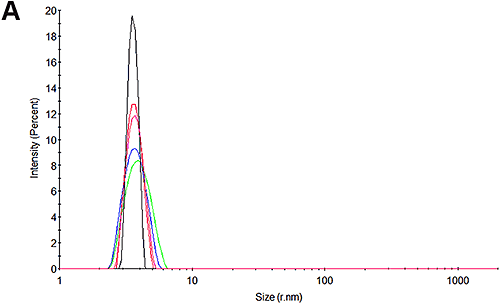
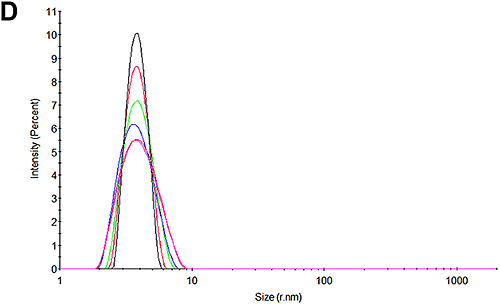
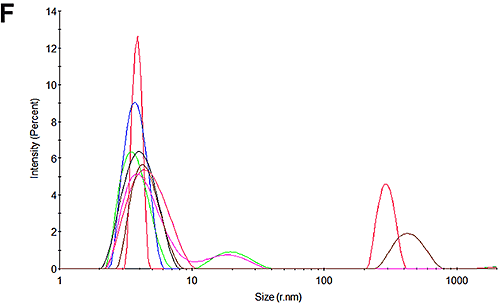
図 3A と 3D は、実験の開始時に pH 7 試料で測定した粒度分布と pH 4.3 試料で測定した粒度分布を示します。 予測どおり、分布にピークが 1 つだけあることがわかります。 pH 7 では 3.8 nm です。これは、HSA でレポートされているものと一致します。 興味深いことに、pH 4.3 ではサイズはやや大きく、4.1 nm です。 これが構造上の変化によるものなのか、高レベルのオリゴマー形成を示すものなのか、凝集の初期兆候を示すものなのかは、この手法では特定できません。
|
|
|
|
|
|
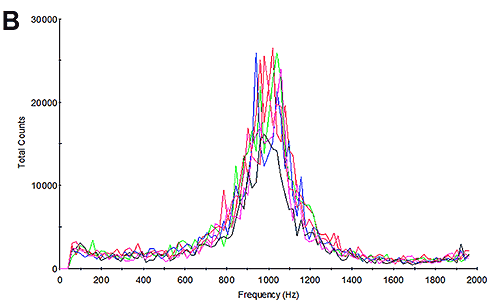
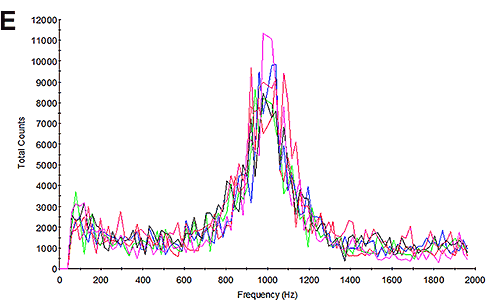
そこで、試料の電気泳動移動度測定を実施し、結果を算出しました。 図3Bと図3E は、電気泳動移動度測定時の周波数プロットを示します。 周波数プロットを調査することは、測定品質の評価に適した方法です [6]。 小さなサイズでは、タンパク質は高速で拡散します。 電気泳動測定中も、電気泳動状態にありますが、高い拡散率は電気泳動では完全に克服できません。 その結果、散乱光の周波数の変化は、明らかに拡散の影響を含む幅広いピークになります。 凝集は大きくなるので、凝集体を含む試料の周波数プロットは、それよりもはるかに小さな拡散成分を持つため、ピークは大きく、シャープなものになります。 これについては、アプリケーションノート MRK1651-02 と参考文献 [4] でより詳細に説明しています。 周波数プロットは、ここでは広く低いピークを持っています。これは、測定結果が主に凝集ではなくタンパク質で構成されていることを示しています。
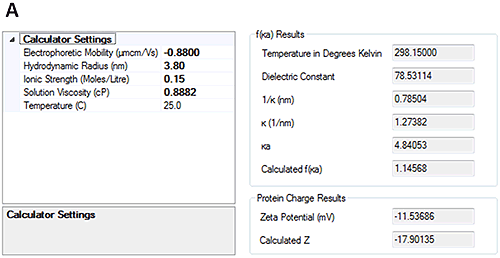
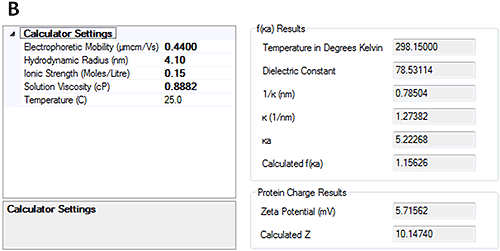
HSA で測定された電気泳動移動度は、pH 7 の場合 -0.88±0.2 µmcm/Vs で、pH 4.3 の場合 0.44±0.2 µmcm/Vs です。 これらの結果は、試料が 2 つの pH ポイントの間の等電点を渡っていることを示しています。
電気泳動移動度を測定すると、ゼータサイザーソフトウェアの新しい解析機能を使用し、測定した流体力学的直径と電気泳動移動度からタンパク質の全般的な電荷を計算できます。 図 4 は、計算された HSA の電荷が pH 7 では約 -18、pH 4.3 では約 +10 であることを示しています。
|
|
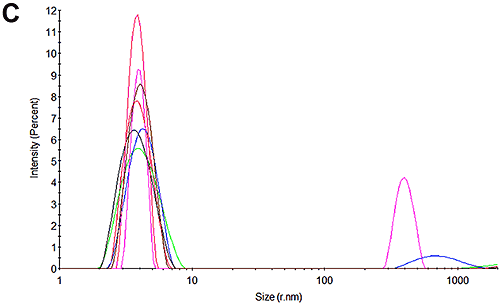
図3Cと図3Fは、電気泳動移動度測定後の試料粒度分布を示しています。 結果は、ごく少量の凝集が測定中に発生していることを示しています。 どちらの場合も、1 次ピークが分布内の大部分を占めており、pH 7では散乱光の 99%、pH 4.3では試料の90%になります。 これらの電気泳動移動度測定の結果には、高い信頼性があるといえます。 pH 4.3 の試料は、時間の経過に従って凝集することが示されています。これは、おそらくpHが低いことが原因です。 この影響は、測定中に起こる少量の凝集の主な原因と考えられます。
測定された電気泳動移動度の結果は、公開されている値と概ね一致しています。 さらに予測どおり、測定されたタンパク質の電気泳動移動度は低いpH値では正の値になります。 結果の正確性に高い信頼性があるため、測定の再現性と品質もすぐれているといえますが、 これは、広い周波数プロットと電気泳動後に確認した粒子径測定で凝集がほとんど発生しなかったことが裏づけられます。
計算されたタンパク質の電荷を Tanford による研究と比較しました [2]。 pH 7では、Tanford は HSA が約 -16 の値を持つと示しています。これは、ここで提示されている値と十分に一致しています。 pH 4.3では、HSAの価数は約+20で、 提示されている値よりもプラス側になります。 これには、実験で使用される緩衝剤やHSAの種類など、複数の理由がありますが、手法の全般的な一致については技術が概ね一致していることは、参考文献 [6] で説明しています。
ゼータサイザー ナノ ZSP と関連ソフトウェアは、拡散障壁手法とともに、信頼性の高い実践的な測定プロセスで高品質の測定を保証します。 これは、次の 3 つの方法で達成されます。
今回、2 mg/mL に過ぎない濃度でも、HSA の電気泳動移動度は異なる緩衝剤で正常に測定されました。 拡散障壁手法では、必要なサンプル量はごく少量で、本文献では20 µLという最低量で測定をし、正しく測定できることが分かりました。
