La stabilité d’une protéine biopharmaceutique joue un rôle déterminant dans le succès ou l'échec du développement d’un produit biopharmaceutique. La stabilité des protéines est un paramètre important lors de l’étape de production, de fabrication, de formulation, de conservation à long terme, d’administration du médicament au patient et pour qu’elles soient efficaces. Les protéines extrêmement stables rencontreront vraisemblablement moins de problèmes pendant le processus de fabrication, ce sont des protéines produites de manière peu onéreuse et qui ont plus de chance de rester fonctionnelles pendant l’étape de formulation et de conservation sans subir d’altération ou d'agrégation. La définition de la qualité par la conception (ou « QbD, Quality by Design ») est une approche du développement de produits biopharmaceutiques dont la caractérisation de la stabilité constitue la première évaluation du potentiel de développement et des propriétés pharmaceutiques d’une molécule candidate potentielle. C’est un concept également utilisé lors de la phase de développement et de fabrication de procédés. Les données relatives à la stabilité sont prises en compte pour déterminer la structure d’ordre supérieur et la cartographie peptidique utilisées à des fins de fabrication, de biocomparabilité et de biosimilarité. Le fait de déterminer la structure d’ordre supérieur d’une protéine devient une pratique de plus en plus courante lorsqu’il s’agit de soumettre une demande d’approbation réglementaire de nouveaux médicaments biopharmaceutiques et de biosimilaires.
Compte tenu de la nature complexe des protéines, les instruments capables de déterminer les propriétés biophysiques ont un rôle important dans l’analyse d’un produit biopharmaceutique. Il existe divers instruments biophysiques généralement utilisés pour évaluer la stabilité d’une protéine, notamment (mais non exclusivement) le dichroïsme circulaire (DC), la diffusion dynamique et statique de la lumière (DLS et SLS), la chromatographie d’exclusion stérique (filtration sur gel) couplée avec la mesure de la diffusion de la lumière multi-angle (SEC-MALS), la spectroscopie infrarouge à transformée de Fourier (FTIR), l'ultrafiltration à flux tangentiel, la chromatographie d’exclusion stérique (SEC), la fluorimétrie différentielle à balayage (DSF), la fluorescence intrinsèque (IF) et la calorimétrie différentielle à balayage (DSC).
Même si toutes ces techniques biophysiques jouent un rôle important dans le développement des produits biopharmaceutiques, il est indispensable d'utiliser la DSC pour déterminer la stabilité thermique. Dans un article datant de 2015 portant sur les techniques biophysiques utilisées pour déterminer la structure d’ordre supérieur d’anticorps monoclonaux, Gokarn et al. ont déclaré : « La DSC reste une technique incomparable pour évaluer la stabilité thermodynamique des protéines dans une solution tampon donnée »[1].
Cet article porte principalement sur l'utilisation de la DSC pour caractériser la stabilité thermique de produits biopharmaceutiques composés de protéines (essentiellement des anticorps), comparer les produits biopharmaceutiques après avoir déterminé la structure d‘ordre supérieur (comparaison par lots, incidence des modifications, etc) et pour développer des biosimilaires.
La DSC est une technique de microcalorimétrie utilisée pour déterminer la stabilité thermique et conformationnelle des protéines, des acides nucléiques, des lipides et d’autres biopolymères[2-7]. La DSC mesure la capacité calorifique en fonction de la température. Les instruments de DSC utilisés pour caractériser une protéine et décrits dans cet article sont des instruments de « compensation de puissance » avec une cellule d’échantillonnage contenant le polymère en solution, placé dans une cellule d'échantillonnage fixe, et une cellule d'étalonnage correspondante contenant le tampon. La capacité calorifique (Cp) d'une cellule d'échantillonnage est comparée à celle d’étalonnage. Au fur et à mesure que la température des cellules augmente, les différences de température relevées entre les cellules d’étalonnage et d'échantillonnage sont systématiquement mesurées et calibrées selon des unités de puissance. La DSC est un « essai de dégradation accéléré » puisque lorsque la protéine est exposée à des températures croissantes, elle se déplie et la capacité calorifique Cp de la protéine augmente (Figure 1).
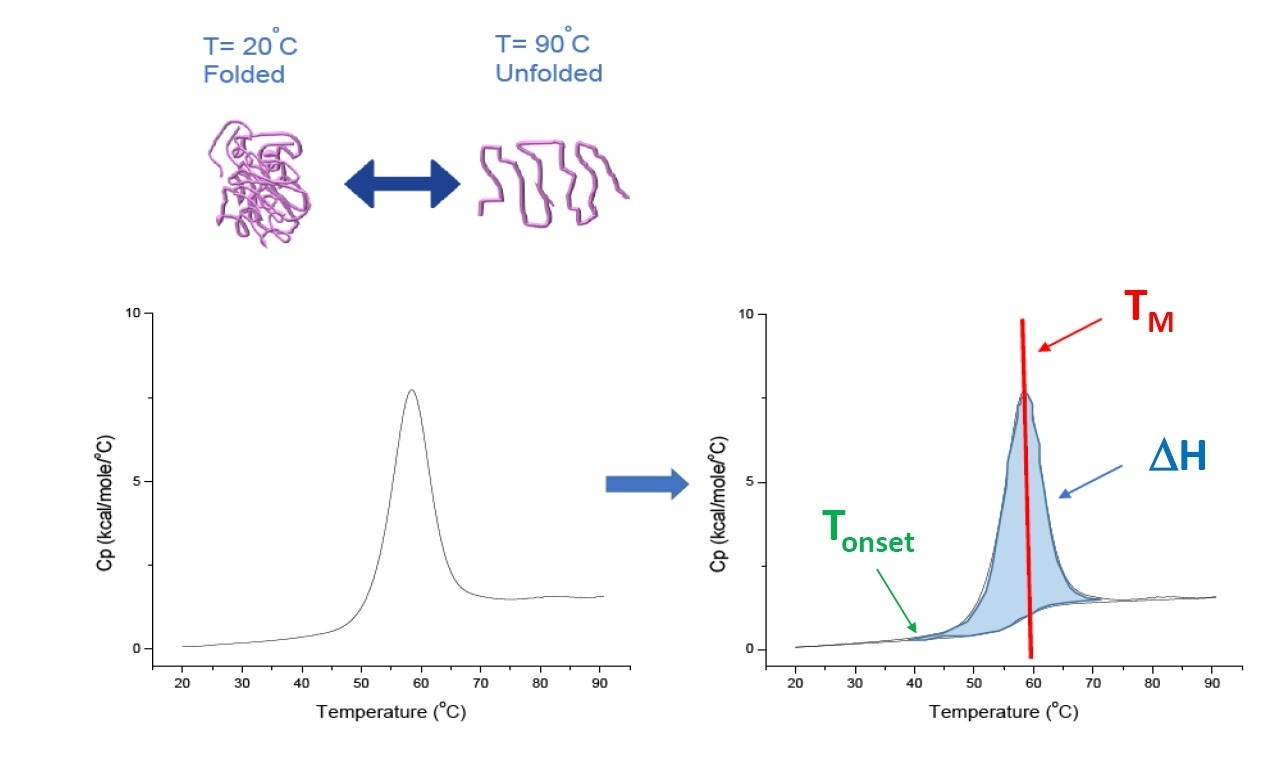
Figure 1: Comprendre le fonctionnement de la DSC. La capacité calorifique (Cp) change lorsque la protéine est dénaturée sous l’effet de la chaleur. Le test DSC débute à une température où la protéine est essentiellement à l’état natif (protéine repliée). Lorsque la température s’élève, la protéine commence à se dénaturer, elle se déplie à certain endroit (Tonset) et la capacité calorifique Cp augmente. La capacité calorifique Cp atteindra sa valeur maximale à une température indiquant un équilibre entre état dénaturé et état natif de la protéine, défini par le point médian de transition thermique ou TM. Pour des valeurs supérieures à cette valeur TM, la protéine sera principalement dénaturée et c’est au terme du test DSC que l’on observera une dénaturation complète de l’ensemble de la protéine. Parmi les paramètres expérimentaux s’appliquant à la DSC, on trouve le point Tonset, le point TM et l’enthalpie de dénaturation (ΔH).
La DSC mesure directement la variation de la capacité calorifique sans avoir recours à des agents fluorescents ou à tout autre sonde ou marqueur. Pour une protéine qui subit une dénaturation réversible, le point médian de transition thermique TM, également appelé température de fusion ou de dénaturation, correspond à la température où la protéine est à son état d’équilibre conformationnel (équilibre entre état natif et dénaturé). Le pic observé sur un thermogramme DSC correspond au point TM.
Le point TM est un bon indicateur de stabilité thermique. Plus ce point est élevé, plus la protéine est stable d’un point de vue thermique. En général, sur un thermogramme DSC plusieurs pics apparaissent avec des protéines multidomaines telles que les anticorps. Par conséquent, il est possible d’identifier différents points TM pour chaque domaine (voir Figure 2).
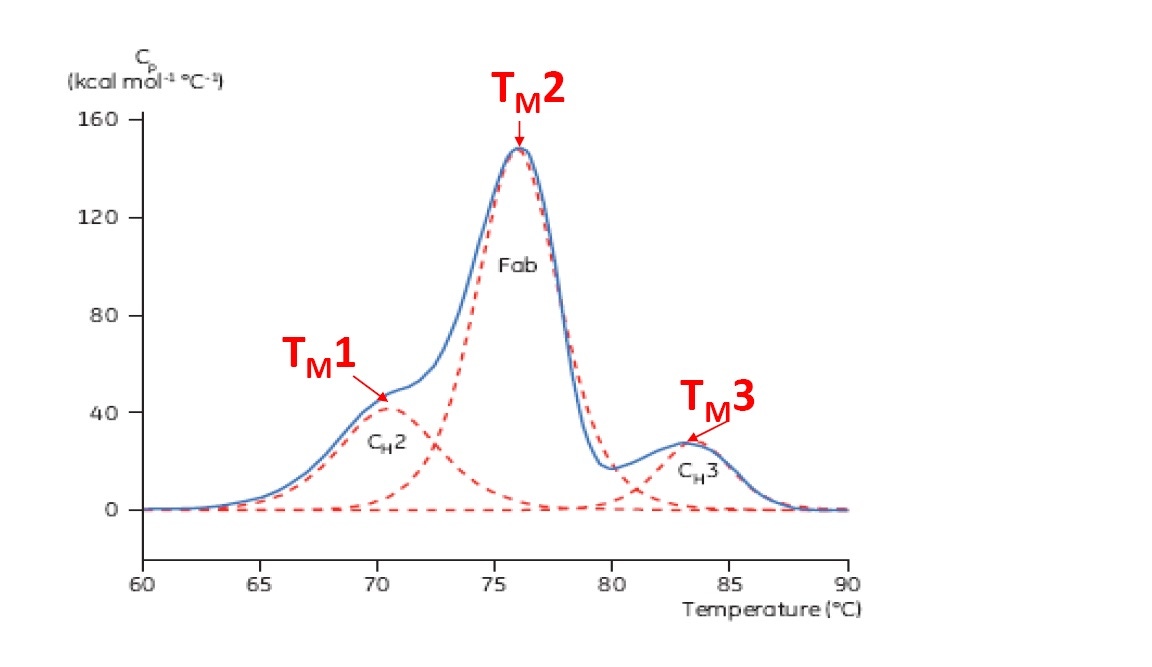
Figure 2 : Thermogramme DSC représentatif d’un anticorps monoclonal constitué d’un domaine CH2, CH3 et d’un fragment Fab. Les lignes rouges en pointillées correspondent aux déconvolutions de pics pour chaque transition de fragments. On observe ainsi les trois points TM.
La DSC fournit d’autres paramètres importants pouvant être utilisés pour caractériser et définir la stabilité protéique, notamment l’enthalpie de dénaturation (ΔH) qui correspond à la surface sous la courbe. La dénaturation de la protéine est un processus endothermique puisque la production d’énergie est nécessaire pour rompre les liaisons secondaires non-covalentes et maintenir la protéine repliée. La DSC détermine également la valeur Tonset (début de la dénaturation), le point ∆Cp (variation de la capacité calorifique associée à la dénaturation) et le point T1/2 (largeur à mi-hauteur du pic, révélateur de la forme du thermogramme représentatif de la dénaturation). L’analyse par DSC permet de définir plusieurs ensembles de paramètres.
La plupart des protéines dénaturées de manière irréversible ont tendance à s’agglomérer ou à cristalliser sous l’effet de la chaleur (dénaturation thermique). Le point TM ainsi que les autres paramètres provenant de l’analyse de protéines dénaturées de manière irréversible réalisée par DSC ne sont pas de vrais paramètres thermodynamiques. Cependant, le classement par ordre de grandeur des valeurs TM établi à partir de l’analyse DSC de protéines dénaturées de manière irréversible réalisée par DSC demeure un précieux paramètre qualitatif du test de stabilité.
Malvern Instruments offre le système DSC MicroCal VP-Capillary[8,9] qui est un calorimètre différentiel à balayage conçu pour analyser le point TM et permettre la caractérisation thermodynamique de protéines et de biopolymères en solution.
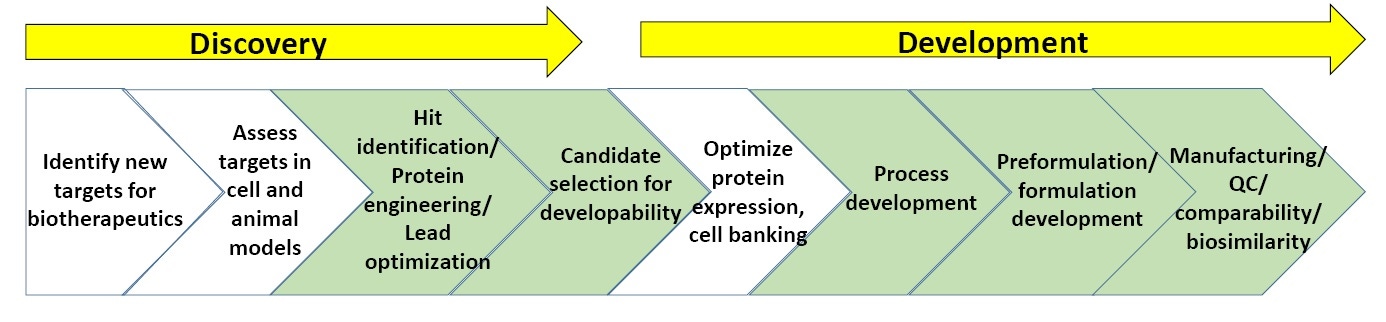
Figure 3: Système général de procédés dans la recherche et le développement de composés biopharmaceutiques
La figure 3 révèle un système général des processus de recherche et développement de composés biopharmaceutiques. Les parties vertes mettent en évidence les points pour lesquels la caractérisation biophysique, notamment les tests de stabilité, sont les plus couramment utilisés (à la fin de cette article, une liste de lecture sur la recherche et le développement de procédés biopharmaceutiques vous est proposée).
Pour développer des produits biopharmaceutiques bénéfiques, les scientifiques cherchent d’abord des molécules présentant une stabilité élevée pendant la sélection du candidat et pensent qu’une stabilité accrue peut être introduite par ingénierie génétique. La purification des protéines est un processus destiné à isoler la protéine d’un milieu dans lequel elle est stable, correctement pliée et active. Il est donc primordial d'utiliser à bon escient les solutions tampon, les additifs, les techniques de purification et les conditions de conservation afin de maintenir une protéine aussi stable que possible à ce stade.
Les protéines-médicaments administrées par voie sous-cutanée doivent être stables et résistantes à des concentrations très élevées en protéine (parfois plus de 100 mg/ml), que ce soit dans un flacon ou une seringue pré-remplie, et cela pendant plusieurs années. Lorsque les molécules de protéines sont exposées à des facteurs de contrainte potentiellement présents lors de la formulation et de la production de procédés biopharmaceutiques comme la chaleur, les produits chimiques, les variations de pH, la pression, cela peut conduire à la dénaturation de la protéine. Les protéines en solution risquent également de subir des modifications telles que la désamidation et l’oxydation pouvant donner lieu à des protéines inactives et dénaturées. Dans le cas de procédés biopharmaceutiques protéiniques, la dénaturation ou d’autres modifications pourraient entraîner la formation d'agrégats susceptibles de réduire leur efficacité ou de leur conférer des propriétés non-fonctionnelles. Il est peut-être encore plus important de souligner que l'agrégation de protéines peut provoquer des réponses indésirables et immunogènes potentiellement mortelles chez un patient. L’utilisation d'une protéine stables comme base biopharmaceutique permet de fabriquer de manière plus avantageuse un produit médicamenteux efficace, bénéfique et sans danger.
La séquence d’acides aminés, ou structure primaire d'une protéine, est un élément de base de la chaîne polypeptidique et de la structure de la protéine. En outre, il est important de comprendre et de caractériser la structure tridimensionnelle de la protéine, également appelée structure d’ordre supérieur. Il existe 3 niveaux de structure d’ordre supérieur de la protéine : la structure secondaire de la protéine qui décrit le repliement local de la structure primaire de la protéine, notamment l'hélice α et le feuillet β, la structure tertiaire qui correspond à la structure tridimensionnelle d’une protéine, provenant d’un ensemble d'éléments de la structure secondaire, et la structure quaternaire qui regroupe l’association d’au moins deux chaînes polypeptidiques identiques ou différentes.
La DSC permet d’analyser la stabilité conformationnelle d’une molécule et les modifications de la structure tertiaire et quaternaire qui surviennent lorsqu’une protéine est dénaturée sous l’effet de la chaleur mais elle permet également de mesurer l’effet des facteurs intrinsèques et extrinsèques sur la stabilité protéique. La DSC est considérée comme la meilleure et la plus performante méthode d’analyse quantitative capable de déterminer la stabilité thermique de protéines biopharmaceutiques. De plus, elle est utilisée comme un bon indicateur de stabilité à long terme[1,10-14]. La DSC permet de générer un point TM qui est un paramètre fréquemment utilisé pour évaluer le niveau de stabilité lors du processus de sélection du produit candidat (potentiel de développement), lors de l’analyse de la formulation et lors du développement de procédés, sachant que la plupart des protéines stables ont un point TM élevé. L’enthalpie (∆H), les valeurs de Tonset, de T1/2 et de ∆Cp mesurées par DSC sont aussi utilisées pour évaluer le niveau de stabilité et valider les données de DSC, l’analyse quantitative de la protéine dénaturée et la structure d’ordre supérieur « cartographie peptidique »[10-14]. .
L’analyse réalisée par DSC d'une protéine présente dans des conditions définies est une analyse quantitative qui peut être reproduite si les protéines analysées sont identiques ou hautement similaires.(Figure 4). En d’autres termes, les thermogrammes DSC présenteront un profil reproductible et les paramètres (notamment les valeurs TM, ΔH et Tonset seront jugés acceptables[12-14]. . Si l’on observe des thermogrammes et des paramètres de DSC différents, cela semblera indiquer qu’il y a eu par exemple soit un repliement anormal de protéines, un phénomène de dégradation, d’agrégation, des écarts observés dans le solvant, des changements dans la modification post-translationnelle ou d’autres modifications de la structure d'ordre supérieur, ayant tous une incidence négative sur la stabilité conformationnelle.
Les résultats reproductibles et quantitatifs font de la DSC un outil clé capable de comparer et déterminer la structure d’ordre supérieur lorsqu’il s’agit d’évaluer un produit pendant sa fabrication (comparabilité par lot et par site) et de comparer les variantes protéiques et les produits modifiés (modifications de structure suite à une glycosilation ou à une oxydation) lorsqu’il s’agit de biosimilarité. Les données de DSC figurent également dans les documents relatifs aux normes en tant qu’éléments importants pour déterminer la structure d’ordre supérieur indispensable à la fabrication de nouveaux procédés et de biosimilaires. Lors d’une enquête réalisée par des chercheurs en technologie biopharmaceutique, la DSC a été définie comme étant une technique d’analyse très utile voire même extrêmement utile pendant la phase de sélection du produit candidat, de développement de la formulation, de caractérisation du produit, de développement de procédés et lorsque l’on parle de comparabilité et de biosimilarité[15].
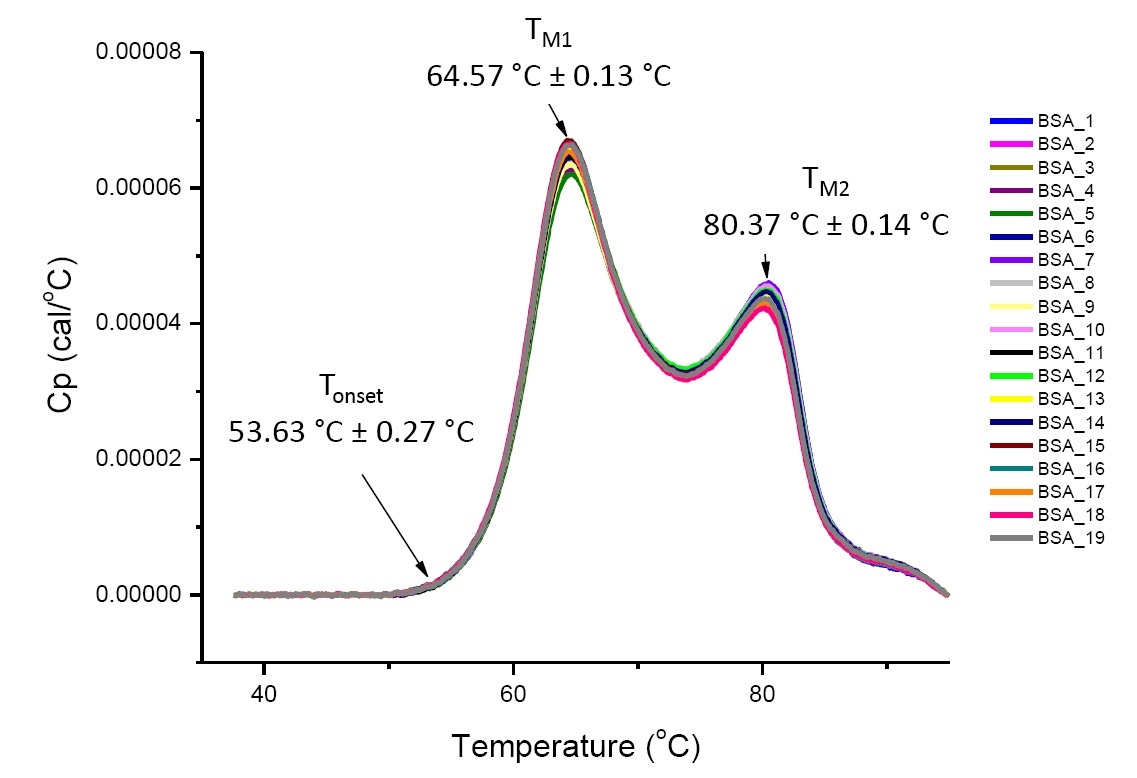
Figure 4 représentant 19 thermogrammes DSC d’albumine de sérum bovin (Sigma A1933, purifié par chromatographie) dans une solution tampon PBS. Les données de DSC qui apparaissent ont été déterminées suite à la normalisation de la fréquence de balayage, au passage d’une solution tampon à une autre et à la suppression d’une solution tampon. La moyenne et l’écart-type des valeurs Tonset, TM1 et TM2 sont indiqués.
Les thermogrammes DSC révèlent l’existence de plusieurs transitions lorsqu’il s’agit de protéines mutidomaines (anticorps) dites dénaturées (voir Figure 2). La DSC est capable d’identifier, de quantifier les différents domaines et de déterminer les TM pour chaque transition. Les valeurs TM sont représentées par les ou les pics de thermogrammes et peuvent être déterminées de manière très simple à partir des données de DSC, sans avoir recours à une analyse complexe des données. Il existe d’autres tests biophysiques capables de déterminer la valeur TM tels que le dichroïsme circulaire (DC), la fluorimétrie différentielle à balayage (DSF) et la fluorescence intrinsèque (IF) mais ils peuvent uniquement mesurer la première valeur TM (rencontrée pour les températures les plus basses) ou la valeur TM la plus prépondérante pour des protéines multidomaines. L’extraction de plusieurs TM obtenues à partir de données spectroscopiques ou fluorescentes nécessite une adéquation complexe des données et peut ne pas être reproductible.
Par rapport à d’autres techniques d’analyse de la valeur TM, la DSC nécessite souvent plus d’échantillons de protéines par test et peut avoir une capacité d’analyse inférieure. Si l’échantillon est limité, il existe une bonne solution qui consisterait tout d’abord à réaliser un classement par ordre de grandeur des TM soit par DSF ou IF puis de sélectionner plusieurs échantillons afin de valider la valeur TM par DSC. Il est important que cette étape de validation des résultats de la valeur TM par DSC soit réalisée et que les techniques de fluorescence ou de spectroscopie ne soient pas considérées comme les seules à pouvoir calculer TM lors des tests de stabilité. Il n'est pas rare dans les tests qui utilisent la fluorescence de voir des artéfacts qui brouillent les mesures et modifient les TM pouvant passer d’une valeur très basse à très haute et inversement. Certaines protéines et solutions tampon ne sont pas compatibles avec la fluorescence, c’est pour cela que le calcul des différences de TM est très compliqué. En définitive, la fluorescence et la spectroscopie sont incapables de mesurer l’enthalpie calorimétrique et de déterminer d’autres paramètres thermodynamiques, contrairement à la DSC qui peut fournir l’ensemble de ces données et permettre d'accéder aux données de stabilité thermique.
La DSC qui est capable de déterminer la stabilité thermique est réputée être un test d’excellence par le secteur biopharmaceutique car cette technique :
Permet de mesurer les écarts thermiques liés à la dénaturation d’une protéine
Permet d’identifier le phénomène de dénaturation d’une protéine, c’est pour cela qu’aucun marqueur, sonde ou puce n’est utilisé. Cela signifie qu’elle ne connaît aucun artéfact contrairement aux tests spectroscopiques ou réalisés par fluorescence, où ils sont fréquents.
S’applique à des protéines natives en solution et peut être utilisée avec la quasi-totalité des tampons et additifs lors de la phase de purification et de formulation des procédés biopharmaceutiques. Certains de ces tampons et additifs sont incompatibles avec la fluorescence et la spectroscopie.
Est facile à configurer et à maîtriser
Permet de contrôler très précisément la température même pour des températures atteignant plus de 130°C. Elle peut donc détecter plus de zones de transitions pour des valeurs TM très élevées. Avec d’autres techniques d’analyse de la valeur TM, les échantillons peuvent être chauffés à des températures atteignant au maximum 100°C, voire inférieures.
Repose sur un test de « dégradation forcée » et n’impose aucune utilisation de tampon pour conserver la protéine avant qu’elle ne soit analysée. La SEC-HPLC et la DLS sont des techniques qui utilisent des échantillons incubés dans des tampons à une température élevée afin de détecter des changements de conformation.
Permet d’extraire facilement les données et est pourvue d’un logiciel intégré capable de les analyser
Peut être utilisée pour détecter les périodes de transitions relatives à la dénaturation et caractériser les protéines multidomaines, les complexes protéiques ainsi que les protéines à domaine unique.
Permet d’accéder à un grand nombre d’informations et fournit des données thermodynamiques en plus de la stabilité conformationnelle et de la valeur TM déterminée.
S'utilise comme principal test pour caractériser la stabilité thermique de procédés biopharmaceutiques, peut être couplée à d’autres outils d’analyse biophysique complémentaires et orthogonaux et permet également de valider d’autres données.
Présente une automatisation à haut débit (système DSC MicroCal VP-Capillary) permettant d’analyser rapidement la stabilité thermique
Comparabilité de produits biopharmaceutiques
Les anticorps monoclonaux et autres médicaments biopharmaceutiques sont des protéines complexes exprimées par des cellules mammaliennes ou bactériennes. Chaque protéine est unique, il faut donc procéder à des recherches approfondies, caractériser et optimiser la protéine de manière précise pour lui permettre de devenir un médicament. Dans le cadre de la recherche et de l’élaboration du médicament, les propriétés de la protéine médicament sont définies, notamment la pureté, l'efficacité et la posologie. Les tests biochimiques, biophysiques et biologiques sont élaborés pour étudier ses caractéristiques et optimisés tout au long du développement du produit.
La caractérisation en détail de la protéine et de la structure d’ordre supérieur (notamment la stabilité) est réalisée lors de chaque opération pour définir les qualités essentielles de la protéine et les paramètres mais également pour contrôler et valider les procédés tout au long du cycle de vie du produit. La définition de la qualité par la conception (ou « QbD, Quality by Design ») est une approche du développement de produits biopharmaceutiques dont la caractérisation de la stabilité constitue la première évaluation du médicament pendant la phase de développement et de fabrication[16,17].
Lors de l’étape de production d’un médicament, la protéine est exposée à des conditions différentes. On trouve notamment :
Différentes solutions, différentes valeurs de pH, différents tampons et différentes concentrations salines
Des additifs de formulation (excipients)
Plusieurs concentrations de protéines
Une alternance répétée de cycles de gel et de dégel, une augmentation de pression et un brassage de protéines
Différents instruments avec lesquels la protéine entre en contact (pompes, membranes d’ultrafiltration/diafiltration, milieu de chromatographie, etc).
Une exposition aux oxydants, aux protéases, au milieu de culture cellulaire, aux variantes de produits, etc.
Toutes ces conditions peuvent perturber les forces et interactions permettant de maintenir une protéine à l’état natif (protéine repliée), ce qui entraîne une dénaturation ou rend la protéine inactive. Une dénaturation chimique due à l’oxydation, à la déamidation, aux changements produits lors de la glycosilation ou à d’autres modifications post-traductionnelles peut également avoir lieu.
Les protéines dénaturées forment souvent des agrégats, ce qui pose problème lors du développement de produits biopharmaceutiques. Les agrégats sont formés de plusieurs protéines monomères et leur formation peut être réversible ou irréversible. On observe des agrégats dont la taille peut être comprise entre un dimère de protéines et de grosses particules visibles à l'œil nu. L'agrégation de protéines réduira tout au moins l’efficacité et la puissance du médicament mais augmentera le coût de production. Les agrégats de protéines et les particules formés soulèvent de grandes préoccupations quant à la possibilité de donner lieu à des réponses immunogéniques, ce qui peut dans des cas extrêmes conduire à la mort du patient. À cause de ce problème, les protéines confrontées au phénomène d'agrégation ont fait l’objet d’un examen plus approfondi de la part de l’industrie biopharmaceutique. Cet examen permet de détecter, de quantifier et de caractériser l’agrégation et les particules lors de l'étape de production et de formulation mais permet également de rechercher des solutions pour réduire ou éliminer l’agrégation de protéines retrouvée dans les produits biopharmaceutiques.
Il est important de montrer que le médicament fabriqué est comparable par sa structure, sa stabilité, sa distribution granulométique, ses techniques biophysiques et fonctionnelles utilisées, à :
La même protéine fabriquée dans des lots antérieurs et dans des lots de référence
La même protéine fabriquée dans différents sites
La même protéine fabriquée à l’aide d’un processus réalisé en amont et en aval
L'intensification du processus réalisé en amont et en aval
La même protéine présente dans une formulation différente
Les tests biochimiques, biophysiques, fonctionnels utilisés à des fins de comparabilité (ou de biocomparabilité) et permettant de déterminer la structure d’ordre supérieur sont conçus pour démontrer que le produit contenant des protéines est « hautement similaire » en termes de qualité par rapport à une protéine de base. Il est indispensable que la structure d’ordre supérieur des médicaments candidats soit attentivement analysée à chaque étape du développement.
Les changements apportés au processus de fabrication d’un produit biopharmaceutique peuvent survenir lors du développement de procédés et suite à l’autorisation de mise sur le marché du médicament. Les raisons de ces changements sont nombreuses :
Amélioration du processus de fabrication (rendement du produit, coûts réduits)
Optimisation d'échelle
Changement d’un site de fabrication
Modifications de la réglementation
Amélioration de la stabilité du produit
Lorsque des changements significatifs sont apportés au processus de fabrication, il faut procéder à un exercice de comparabilité pour évaluer l’incidence de ces changements sur la qualité, l’innocuité et l’efficacité du produit biopharmaceutique. Le fait d’avoir recours à la comparabilité ne signifie pas pour autant que les aspects qualitatifs du produit figurant avant et après les changements soient identiques mais plutôt qu’ils soient hautement similaires. Les changements apportés au processus de fabrication ne devraient avoir aucun effet néfaste sur la qualité du produit.
La comparabilité est une source importante de préoccupations lorsqu’il s’agit de produits thérapeutiques à base de protéines, et est examinée par plusieurs organismes internationaux de réglementation[18,19,20]. Aucune méthode analytique particulière ne peut être utilisée pour évaluer la comparabilité de protéines médicaments. Les études de comparabilité et de structure d’ordre supérieur comprennent (mais ne se limitent pas à) : la DSC, la diffusion dynamique de la lumière (DLS), la fluorescence, le dichroïsme circulaire (DC), la chromatographie d’exclusion stérique (SEC), l'ultrafiltration à flux tangentiel, la spectroscopie Raman, la technologie « Nanoparticle Tracking Analysis » (NanoSight de Malvern Instruments) qui détecte et visualise des populations de nanoparticules, la technique de la mesure de masse résonnante (Archimedes de Malvern Instruments) et la microscopie. La bioactivité et l'efficacité de protéines sont également contrôlées par des tests biologiques, la calorimétrie de titration isotherme (ITC) et la résonance plasmonique de surface (SPR)
Les informations transmises par chaque méthode sont spécifiques et lorsqu’elles sont analysées conjointement, elles permettent de fournir des éléments importants en vue de caractériser la structure d’ordre supérieur et à des fins de comparabilité. L’architecture de la structure d’ordre supérieur permet d’assurer la cohésion dans la structure de la protéine pendant le cycle de développement du procédé, pendant la phase de comparabilité par lots, lorsque les procédés sont transférés vers un nouveau site de fabrication et lorsque la production est modifiée. Les informations issues de la caractérisation de la structure d’ordre supérieur sont également indispensables à l’évaluation de biosimilaires.
Il est important de souligner que l’évaluation et l’instauration de la comparabilité dans le processus de fabrication n’a rien à avoir avec l’évaluation réalisée pendant le développement d’un médicament biosimilaire. Il faut savoir que le développement d’un biosimilaire est un processus bien plus complexe. Étant donné qu’un biosimilaire n’est généralement pas produit par la même société qui a développé le médicament constitué de la molécule mère (également appelé produit de référence), le développement de biosimilaires s’appuie sur des techniques d'ingénierie inverse pour mettre en place des processus en amont et en aval. Le fabriquant de biosimilaires doit s’aligner sur la qualité du produit (biosimilarité) concernant un petit nombre de produits commerciaux de référence, ce qui demande de recueillir d’avantage d'éléments biophysiques et biochimiques pour démontrer la biosimilarité par rapport à ce qui est généralement pratiqué lors de la fabrication.
Étant donné que la DSC fournit des informations sur la stabilité thermique d’une protéine présente dans des solutions différentes de solvant et que les résultats de DSC sont reproductibles (Figure 4), on la retrouve dans les études de comparabilité pour montrer que le processus n’a pas d’effet sur la stabilité du produit et que les produits fabriqués issus de différents lots et/ou de différents sites de fabrication sont hautement similaires.
Jiang et Nahri[21] ont examiné plusieurs techniques biophysiques à des fins de comparabilité pendant le développement et la fabrication de procédés. Il ont mentionné l’utilisation de la DSC comme un des instruments biophysiques de caractérisation employé pour comparer un médicament à base d’anticorps monoclonaux exprimés par deux lignées cellulaires différentes (lignée cellulaire 1 et 2), ayant connu des modifications dans le processus de fabrication (processus 2pA et 2pB). Les trois produits à base de protéines qui en résultent ont été évalués par spectroscopie infrarouge à transformée de Fourier (pour comparer la structure secondaire), par mesure du dichroïsme circulaire dans l’UV proche et par fluorescence intrinsèque (pour comparer la structure tertiaire), par fluorescence du complexe 8-anilino-1-naphthalene sulfonate (pour comparer l’hydrophobicité de surface), par diffusion dynamique de la lumière (pour comparer les propriétés hydrodynamiques et la granulométrie des protéines) et par DSC (pour comparer la stabilité thermique et la solubilité).
Les résultats de ces tests biophysiques ont démontré que les structures secondaires et tertiaires des trois échantillons étaient similaires. Les résultats de DSC ont indiqué une augmentation significative de la stabilité thermique de la protéine issue de la lignée cellulaire 2pB par rapport à l’échantillon 2pA et à l’échantillon issu de la lignée cellulaire 1. L’échantillon 2pA et l’échantillon issu de la lignée cellulaire 1 semblaient plus hétérogènes d’après la DSC. En général, on constate 2 à 3 transitions pour les anticorps (voir figure 2 représentant un thermogramme DSC « type » d’un anticorps). D’après l’analyse par DSC de ces échantillons, on a observé la présence d’un grand nombre de transitions thermiques qui se chevauchent, ce qui indique qu’il existe une hétérogénéité des échantillons. Les résultats de DSC ont indiqué que les modifications observées dans le processus permettaient d’améliorer l’homogénéité et la stabilité de l’anticorps monoclonal. Selon les données de comparabilité relatives à l’analyse fonctionnelle, biochimique et biophysique présentées dans l'article, la lignée cellulaire 2 et le processus 2pB ont été considérés comme capables de produire la protéine la plus stable[21].
Jiang et Nahri[21] ont également évoqué l’utilisation de la DSC pour étudier la comparabilité d’un produit à base de protéines fabriqué dans différents sites de fabrication. La protéine Y était composée de deux chaînes polypeptidiques constituées de monomères et liées par des ponts disulfure à la région du fragment Fab de la molécule. Au cours de son développement, la protéine Y était fabriquée dans différents sites. Pour s'assurer que la protéine présente dans ces différents lots était similaire en termes de conformation (retrouvée à l’état natif), de structures secondaires et tertiaires, de stabilité thermique et de granulométrie, quatre échantillons différents de la protéine Y, représentant 3 échantillons obtenus dans différents sites et celui obtenu à partir de la protéine de référence, ont été analysés par spectroscopie infrarouge à transformée de Fourier, par mesure du dichroïsme circulaire dans l’UV proche et lointain, par fluorescence, par DSC et par ultrafiltration à flux tangentiel.
Les analyses de DSC de l’étalon de référence et des trois échantillons issus de différents sites de fabrication ont montré que les deux transitions thermiques de chaque échantillon et les quatre profils DSC étaient identiques dans le cadre de l’étude de la variabilité expérimentale. Cela semble indiquer qu’aucune différence de stabilité thermique des échantillons de protéines n’a été relevée et qu’elles étaient toutes sous leur forme native correctement repliée. Les résultats combinés de caractérisation biophysique, notamment ceux obtenus par DSC, ont démontré que les quatre échantillons de la protéine Y étaient similaires, présentaient une structure secondaire et tertiaire adéquate et étaient homogènes[21].
Si les thermogrammes DSC n’avaient pas été identiques, cela aurait pu indiquer une éventuelle modification post-translationnelle ou une dénaturation chimique ayant eu lieu dans la protéine. Arthur, et al.[22] ont démontré que la sensibilité de la DSC pour détecter les changements de structure d’ordre supérieur liés à la stabilité provenait de la réaction d'oxydation, une voie de dégradation chimique très souvent observée. Si un produit biopharmaceutique est altéré par l’oxydation pendant l’étape de purification ou de conservation lors de la formulation finale, cela peut nuire à son efficacité et même augmenter le risque de formation d'agrégats. Lors de cette étude, les produits constitués de protéines obtenus à partir de trois classes structurales ont été évalués à plusieurs niveaux d’oxydation. On a observé pour chaque protéine une diminution linéaire de la valeur TM en fonction de l’oxydation de la méthionine. Les auteurs ont également observé des différences dans le taux de variation de la valeur TM ainsi que des différences de stabilité du domaine de la valeur TM entre classes structurales et au sein des classes structurales[22]. En revanche, le dichroïsme circulaire dans l’UV proche et la spectroscopie de fluorescence étaient bien moins sensibles aux changements de conformation induits par l’oxydation. La DSC est une méthode de caractérisation structurale plus appropriée pour observer l’oxydation des protéines par rapport à ces méthodes de spectroscopie[22]. Lors de cette étude, on s’est intéressé à la protéine IgG2B et on a détecté par DSC des variations de TM précédant une perte d’efficacité relative, ce qui prouve que la DSC est le principal indicateur d’une affinité de liaison réduite entre l'antigène et l’anticorps. La spectroscopie de masse permet de détecter l’oxydation de la méthionine qui se produit à des niveaux d’oxydation inférieurs à ceux ayant un impact fonctionnel ou provoquant un changement de conformation. En s’appuyant sur les changements de TM observés par DSC et par spectroscopie de masse, il est possible de déterminer de manière fiable la relation qui existe entre une modification de la structure primaire, les changements de structure d’ordre supérieur, les variations de stabilité conformationnelle et l’impact fonctionnel.
Morar-Mitrica et al.[12] décrivent plusieurs études de cas faisant appel à la DSC pour caractériser la structure d’ordre supérieur et des études de comparabilité de produits biopharmaceutiques. Ils s’intéressent à la caractérisation de l’oxydation d’anticorps monoclonaux détectable par les variations de TM (similaires à celles décrites dans la référence 22), aux profils de thermogrammes DSC reproductibles, à la mesure de la valeur TM, de la valeur T1/2, de la valeur Tonset et de ∆H utiles pour comparer des produits biopharmaceutiques et à la caractérisation plus approfondie par DSC d’anticorps monoclonaux glycosylés présentant différents niveaux de glycosylation (une modification post-translationnelle souvent observée).
Shahrokl et al.[23] expliquent l’utilité de la DSC lors des études de comparabilité dans le cadre d’une demande d’autorisation de mise sur le marché de produits biologiques, soulignant la simplicité d'utilisation de la DSC et de ses données. Les auteurs décrivent la façon dont était utilisée la DSC pour comparer une glycoprotéine d'un poids moléculaire de 51 kDa avant et après la survenue d’un changement dans le processus de fabrication. Ce changement a été mis en œuvre pour accroître le rendement de la production et pour supprimer les substances d’origine animale retrouvées dans le milieu de culture cellulaire. En utilisant la DSC, les auteurs ont pu évaluer trois lots de glycoprotéines produits grâce à chaque opération de fabrication et observer la superposition des thermogrammes DSC qui présentaient une seule transition à 60,7 °C +/- 0,1°C. Les résultats de DSC ont montré que le changement apparu dans le processus de fabrication n'affectait pas la stabilité conformationnelle de la glycoprotéine[23].
Lubiniecki et al.[24] ont évalué l’incidence de ces changements sur la structure et l’activité de l’anticorps au cours du développement du produit en réalisant trois études de comparabilité portant sur deux anticorps IgG1 monoclonaux candidats différents (anticorps A et B). Deux d’entre elles ont intégré la DSC comme instrument analytique utilisé en biochimie et biophysique. L’une de ces études a examiné l’extrapolation du procédé et le transfert vers le site de fabrication, accompagné du passage sous forme liquide d’une forme galénique lyophilisée. Les thermogrammes DSC représentatifs des formulations lyophilisées et liquides des anticorps A et B ont indiqué une bonne corrélation[24]. La DSC, utilisée en parallèle avec la spectroscopie de masse (MS), le dichroïsme circulaire (CD), la chromatographie d’exclusion stérique (SEC) et d’autres techniques, a indiqué que le changement apporté à la fabrication n’avait pas de répercussion sur la structure ou la fonction des anticorps.
La troisième étude de comparabilité a permis d’analyser la formulation sous forme liquide de l’anticorps présent dans des seringues des flacons pré-remplis[24]. Les résultats obtenus à partir de la DSC, du CD, de la SEC et d’autres techniques semblent indiquer une structure moléculaire, une activité biologique et des profils de dégradation similaires. L’existence d’une légère (quoique significative) augmentation des taux de particules imperceptibles dans les seringues pré-remplies constitue la seule exception notable[24].
Un produit biosimilaire (aussi appelé « follow on biological » aux États-Unis ou « produit biologique ultérieur » au Canada) est un produit biopharmaceutique qui a été approuvé par un organisme de réglementation en fonction de sa similarité avec un produit biologique déjà approuvé, dénommé produit de référence (considéré également comme le produit contenant la molécule mère). Depuis des années, les biosimilaires sont disponibles dans le monde[25-29]. Au moment où cet article a été rédigé (octobre 2016) la FDA a approuvé quatre biosimilaires et l’agence européenne pour l’évaluation des médicaments (EMEA) en a approuvé 19.
Il faut souligner que le développement d’un biosimilaire n’est pas comparable au développement d’un médicament générique constitué de petites molécules. Les petites molécules thérapeutiques sont chimiques et leur synthèse suit un processus contrôlé. Étant donné que les produits biopharmaceutiques sont habituellement des protéines, il existe un certain degré de variation dans la teneur en protéine, y compris entre les différents lots du même produit, en raison de la variabilité intrinsèque du système biologique d’expression et du processus de fabrication. Parmi ces variations, on observe des différences de modification post-translationnelle et des différences de structure d’ordre supérieur. En raison du poids moléculaire élevé des protéines, de leur structure complexe et des variations naturelles, les biosimilaires ne sont pas aussi simples à produire que les génériques. Un biosimilaire n’est pas un doublon du produit de référence et c'est la raison pour laquelle il est qualifié de « similaire » ou de « hautement similaire » au produit de référence.
Le développement d’un biosimilaire consiste à comparer les propriétés physico-chimiques, analytiques et fonctionnelles de la protéine référence et du biosimilaire. Ces tests sont complétés par des données comparatives cliniques et non-cliniques pour démontrer l’efficacité et l’innocuité du produit. Avant d’être approuvé, il convient de vérifier que le biosimilaire ne présente aucune différence clinique significative en termes de sécurité et d’efficacité par rapport au produit de référence. Seules quelques légères différences de composants inactifs cliniquement sont retrouvées dans les produits biosimilaires. On s’attend à ce que le biosimilaire fonctionne exactement de la même manière que le produit de référence, c’est à dire qu’il soit approuvé pour les mêmes usages.
Les agences de pharmacovigilance évaluent les biosimilaires en fonction de leur degré de similarité avec leurs produits de référence. Étant donné la complexité des produits biopharmaceutiques, il est peu probable que deux fabricants différents puissent produire deux produits identiques, même si des systèmes d’expression hôtes, des procédés, des techniques identiques sont utilisés. Par conséquent, les fabricants de biosimilaires doivent s’appuyer sur des études de comparabilité et des analyses de la structure d’ordre supérieur, tel que décrit précédemment[30,31]. La nécessité de disposer d’information analytique et la volonté de respecter des délais réduits pour le développement de biosimilaires demande à ce que soit appliqué à chaque étape, et plus particulièrement lors de la fabrication, un module de comparabilité avec la molécule de référence.
Les biosimilaires doivent être obtenus par « rétro-ingénierie » pour être le plus possible semblables au produit de référence. La caractérisation analytique d’un biosimilaire comprend l’évaluation de la structure primaire, secondaire, tertiaire et quaternaire, de l’activité biologique et de l’analyse des impuretés de fabrication et du produit. La DSC est en général utilisée comme instrument biophysique pour analyser la structure d’ordre supérieur et pour démontrer qu’un biosimilaire présente un profil DSC hautement similaire, des valeurs TM, Tonset et autres paramètres thermodynamiques similaires par rapport au produit de référence.
Trois des quatre biosimilaires approuvés par la FDA (depuis octobre 2016) ont permis d’inclure la DSC dans le dossier de présentation du médicament comme un des tests d'analyse de la structure d’ordre supérieur. Parmi ces trois biosimilaires, on trouve :
Erelzi (de la filiale Novartis Sandoz) qui est un biosimilaire d’Enbrel (étanercept), médicament du groupe américain Amgen[32]
Inflectra (de Celltrion), dénommé Remsima sur d’autres marchés, qui est un biosimilaire de Remicade (infliximab), médicament de la société pharmaceutique belge Janssen[33,34]
Amjevita (de Amgem) qui est un biosimilaire de Humira (adalimumab), médicament de l’entreprise pharmaceutique américaine Abbvie[35,36]
Sinha-Datta et al.[37] ont démontré que l'utilisation de la DSC (DSC MicroCal VP-Capillary) et de la résonance plasmonique de surface (SPR) permettait de comparer deux anticorps monoclonaux thérapeutiques (mAb1-i et mAb2-i) avec leurs biosimilaires (mAb1-B et mAb2-B1, B2, B3) en fonction de leur stabilité thermique, cinétique et de leur affinité. Ils ont observé que les biosimilaires étaient hautement similaires à leurs échantillons de référence par rapport à leur activité biologique illustrée à partir des résultats obtenus par SPR. La DSC a permis une fois encore de confirmer l’existence de biosimilarité entre produits de référence et biosimilaires. L‘analyse réalisée par DSC a révélé une grande similarité structurale en ce qui concerne les anticorps présents dans le produit de référence et dans le biosimilaire avec une valeur TM principale de 84,1°C (pour mAb1) et de 72,8°C (pour mAb2).
Les informations présentées dans cet article illustrent clairement l’importance et l’efficacité d’appliquer la DSC en tant que test biophysique de stabilité à des fins de comparabilité biopharmaceutique et de caractérisation de biosimilarité. L’interprétation des résultats issus de la DSC et d’autres tests biophysiques et biochimiques a permis aux sociétés biopharmaceutiques de prendre des décisions éclairées sur la stabilité de la protéine et la comparabilité pendant la phase de production, de veiller à ce que chaque lot de protéine soit hautement similaire au lot de référence et que tout changement apporté au processus de fabrication n’ait pas d’incidence sur la stabilité conformationnelle de la protéine. La DSC est également considérée comme un test d’analyse de la structure d’ordre supérieur lorsqu’il s’agit du développement de biosimilaires puisqu’elle permet de démontrer que le médicament biosimilaire est « hautement similaire » au produit de référence.
Analytical Techniques for Biopharmaceutical Development, R. Rodriguez-Diaz, T. Wehr, S. Tuck (eds.), Taylor & Francis, New York, NY, USA (2005).
Biophysical Characterization of Proteins in Developing Biopharmaceuticals, D.J. Houde, S.A. Berkowitz (eds.), Elsevier, Amsterdam, Netherlands (2015).
Biophysical Methods for Biotherapeutics: Discovery and Development Applications, T.K. Das (ed.) John Wiley & Sons, Hoboken, NJ, USA (2014).
Biophysics for Therapeutic Protein Development, L.O. Nahri (ed.), Springer, New York, NY, USA (2013).
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1176 (2014). doi: 10.1021/bk-2014-1176.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1201 (2015). doi: 10.1021/bk-2015-1201.
State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 3. Defining the Next Generation of Analytical and Biophysical Techniques, J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1202 (2015) DOI: 10.1021/bk-2015-1202.
1. Gokarn, Y., Agarwal, S., Arthur, K., et al., Biophysical Techniques for Characterizing the Higher Order Structure and Interactions of Monoclonal Antibodies, in: State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study. J.E. Schiel, D.D. Davis, O.V. Borisov (eds.), ACS Symposium Series Vol 1201, American Chemical Society, Washington DC, USA, pages 285-327 (2015).
2. Cooper, A., Nutley, M. A., and Wadood, A., Differential Scanning Calorimetry, in: Protein-Ligand Interactions: Hydrodynamics and Calorimetry, A Practical Approach, S.E. Harding, B.Z. Choudry (eds). Oxford University Press, Oxford, UK, p. 287-318 (2001).
3. Malvern Instruments Whitepaper “Differential Scanning Calorimetry (DSC) Theory and Practice” http://www.malvern.com/en/support/resourcecenter/Whitepapers/WP140701-dsc-theory-and-practice.aspx.
4. Bruylants, G., Wouters, J., and Michaux, C., Current Med. Chem. 12, 2011-2020 (2005) doi: 10.2174/0929867054546564.
5. Jelesarov, I., and Bosshard. H.R., J. Mol. Recognit. 12, 3-18 (1999) doi: 10.1002/ (SICI)1099 1352(199901/02)12:1<3:AID-JMR441>3,0.CO; 2-6.
6. Choi, M.H., and Prenner, E.J., J. Pharm. Bioallied Sci. 3, 39-59 (2011) doi: 10.4103/0975-7406,76463.
7. Johnson, C.M., Arch. Biochem. Biophys. 531, 100-109 (2013) doi: 10.1016/j.abb.2012,09.008.
8. Plotnikov, V., Rochalski, A., Brandts, M., Brandts, J.F., Williston, S., Frasca, V., and Lin, L.N., Assay Drug Devel. Technol. 1, 83-90 (2004) doi:10.1089/154065802761001338.
9. www.malvern.com\fr
10. Demarest, S.J., and Frasca. V., Differential Scanning Calorimetry in the Biopharmaceutical Sciences, in: Biophysical Characterization of Proteins in Developing Biopharmaceuticals, D.J. Houde, S.A. Berkowitz (eds.), Elsevier, Amsterdam, Netherlands, p. 287-306 (2015).
11. Remmele, R.L., Microcalorimetric Approaches to Biopharmaceutical Development, in: Analytical Techniques for Biopharmaceutical Development, R. Rodriguez-Diaz, T. Wehr, S. Tuck (eds.), Taylor & Francis, New York, NY, USA, p. 327-381 (2005).
12. Morar-Mitrica, S., Nesta, D., and Crofts, G., BioPharm Asia 2, 46-55 (2013).
13. Kirkitadze, M., Hu, J., Tang, M., and Carpick, B., Pharm. Bioprocess. 2, 491-498 (2014) doi: 10.4155/PBP.14,27.
14. Wen, J., Arthur, K., Chemmalil, L., Muzammil, S., Gabrielson, J., and Jiang, Y., J. Pharm. Sci. 101,955-964 (2012) doi: 10.1002/jps.22820.
15. Gabrielson, J.P., and Weiss, W.F., J. Pharm. Sci. 104, 1240-1245 (2015) doi: 10.1002/jps24393.
16. Cooney, B., Jones, S.D., and Levine, H., BioProcess Int. 14(6), 28-35 (2016). http://www.bioprocessintl.com/analytical/upstream-development/quality-by-design-for-monoclonal-antibodies-part-1-establishing-the-foundations-for-processdevelopment/
17. Cooney, B., Jones, S.D., and Levine, H., BioProcess Int. 14(8), 24-33 (2016). http://www.bioprocessintl.com/2016/quality-design-monoclonal-antibodies-part-2-process-design-space-control-strategies/
18. http://www.who.int/biologicals/biotherapeutics/rDNA_DB_final_19_Nov_2013.pdf
19 http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003935.pdf
20. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM496611.pdf
21. Jiang, Y., and Nahri, L.O., J. Am. Pharm. Rev. 9, 34-43 (2006).
22. Arthur, K.K., Dinh, N. , and Gabrielson, J. P., J. Pharm. Sci. 104, 1544-1554 (2015) doi: 10.1002/jps.24313.
23. Shahrokh, Z., Salamat-Miller, N., and Thomas, J.J., Biophysical Analyses Suitable for Chemistry, Manufacturing, and Control Sections of the Biologic License Application (BLA), in: Biophysical Methods for Biotherapeutics: Discovery and Development Applications, T.K. Das (ed.) John Wiley & Sons, Hoboken NJ USA pages 317-353 (2014).
24. Lubiniecki, A., Volkin, D.B., Federici, M, et al., Biologicals 39, 9-22 (2011). doi: 10.1016/j.biologicals.2010.08.004
25.http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/
26.http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
27.http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf
28 Bas, T.G., and Oliu Castillo, C., Biomed Res Int. 2016, 5910403 (2016). doi: 10.1155/2016/5910403.
29. Tsuruta, L.R., Lopes dos Santos, M., and Moro, A.M., Biotechnol Prog. 31, 1139-1149 (2015). doi: 10.1002/btpr.2066
30 Kálmán-Szekeres, Z., Olajos, M., and Ganzler, K., J. Pharm. Biomed. Anal. 69, 185-195 (2012). doi: 10.1016/j.jpba.2012.04.037
31 Berkowitz, S.A., Engen, J.R., Mazzeo, J.R., and Jones, G.B., Nat. Rev. Drug Discov. 11, 527-540 (2012). doi: 10.1038/nrd3746
32.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM510493.pdf
33.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM484860.pdf
34. Jung, S.K., Lee, K. H., Jeon, J. W., et al., MAbs 6, 1163-1177 (2014). doi: 10.4161/mabs.32221
35.http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ArthritisAdvisoryCommittee/UCM510293.pdf
36. Liu, J., Eris, T., Li, C., Cao, S., and Kuhns, S., BioDrugs 30, 321-338 (2016). doi: 10.007/s40259-016-0184-3.
37. Sinha-Datta, U., Khan, S., and Wadgaonkar, D., Biosimilars 5, 83-91 (2015) doi: 10.2147/BS.S85537 https://www.dovepress.com/label-free-interaction-analysis-as-a-tool-to-demonstrate-biosimilarity-peer-reviewed-article-BS
