Ce livre blanc aborde les thèmes principaux concernant l'analyse par DSC, en l'illustrant à l'aide d'applications élémentaires. The benefits and specifics of DSC are discussed.
L'avantage principal de la méthode DSC est qu'elle repose sur des mesures de la chaleur et permet donc la caractérisation des biomolécules natives. En outre, en raison de l'absence de mesures spectroscopiques, il n'est pas indispensable d'utiliser des échantillons clairs sur le plan optique. Par ailleurs, la caractérisation ne se limite pas à la température de fusion (Tm), mais permet également d'obtenir des données sur les forces participant au repliement des biomolécules et les mécanismes de dénaturation.
Les propriétés thermodynamiques des macromolécules biologiques dans les solutions ne sont pas faciles à interpréter de manière isolée. Les valeurs mesurées correspondent toujours à l'effet net des interactions entre des groupes au sein de la molécule et des interactions entre ces mêmes groupes et le solvant. Les systèmes biologiques équilibrant les rapports énergétiques représentent donc la différence entre des interactions favorables et celles qui sont potentiellement défavorables. Cette observation constitue pour de nombreux chercheurs une révélation, dont les mesures par DSC prouvent clairement le bien-fondé. Cependant, il n'y a aucun lieu de se décourager, car une approche minutieuse et méthodique peut permettre de démêler la complexité de l'interaction. La DSC présente de nombreuses autres applications, reposant en partie sur l'aptitude à observer l'intégralité des phénomènes thermodynamiques associés aux changements dans les forces régissant la stabilisation de la macromolécule et sur l'absence de composants optiques. La DSC peut mesurer l'influence d'effets très simples liés à la protonation des macromolécules sur les propriétés thermodynamiques et la stabilité. Cette approche peut être étendue à tout ligand présentant des liaisons non covalentes, ce qui confère un caractère polyvalent à cette méthode d'analyse des liaisons. Dans des circonstances favorables, cette approche permet de déterminer les constantes de liaison. Nous allons étudier ici les principes thermodynamiques des mesures DSC, ainsi que les applications de cette méthode dans l'étude de la stabilité des protéines, aussi bien indépendamment que dans le cadre d'interaction avec des ligands. Cependant, les techniques sont tout à fait transférables à d'autres macromolécules biologiques, telles que des acides nucléiques, lipides et ainsi de suite.
Lorsque la température augmente au-delà d'un seuil physiologique normal, les protéines subissent une transformation, passant d'une conformation structurée, native et biologiquement active (N) à une conformation déstructurée, dénaturée et inactive (D). Les protéines partagent ce comportement avec d'autres macromolécules biologiques (ADN, lipides, etc.) et avec des polymères organiques en règle générale. En suivant les signaux émanant de la protéine lors de cette transition de conformation, il est possible d'obtenir une courbe sigmoïde conforme à l'image ci-dessous.
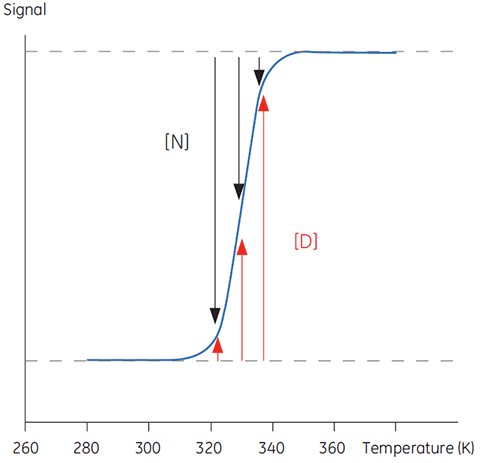
|
Une fois que la structure de la protéine a fondu de la sorte, aucun changement ne se produit concernant la nature covalente de la molécule. Seules les interactions non covalentes sont perturbées. Dans bien des cas, après refroidissement, la protéine reprend spontanément sa forme initiale, en obéissant à la conformation active native. Les statuts N et D forment donc un équilibre réversible, où la température joue le rôle de variable intensive :

|
En Figure 1, les proportions de N et D évoluent à mesure que l'équilibre se dirige vers D, en s'accompagnant d'une augmentation de la température. À toute température, il est possible de définir une position d'équilibre, la constante d'équilibre (Keq), qui reflète simplement les concentrations relatives de N et D. Sur une échelle logarithmique, cette constante d'équilibre s'exprime sous forme d'énergie libre de Gibbs (ΔG) :

|

|
où R est la constante des gaz et T la température en degrés Kelvin. La température à laquelle les concentrations de D et N sont égales se définit comme le point médian de la transition ou température de fusion Tm. À cette température, Keq est égale à 1 tandis que ΔG est nulle. La valeur Tm constitue un paramètre important pour toute protéine, car elle dénote sa stabilité thermique. Sous cette température, la concentration en protéine native est plus élevée, tandis qu'au-dessus de Tm, la part dénaturée de la protéine est majoritaire.
Ce comportement de fusion chez les protéines provient du fait que leurs structures natives sont stabilisées par ces nombreuses interactions, elles-mêmes dépendantes de la température. La stabilisation par enthalpie (ΔH) nécessite diverses interactions, notamment la création de liaisons, leur formation de structure et leur réduction en énergie interne. La stabilisation par entropie (ΔS) reflète les interactions de détérioration de l'ordre et augmente le nombre de modes d'organisation du système avec la même énergie. Ces termes sont liés à la valeur ΔG dans l'équation bien connue :

|
En combinant les équations 3 et 4, et en modifiant leur agencement, on peut élaborer l'Équation 5 de van't Hoff, qui permet de représenter la variation de la constante d'équilibre afin d'obtenir l'enthalpie et l'entropie de la dénaturation thermique sous forme d'une relation linéaire simple (lnKeqen fonction de 1/T) :
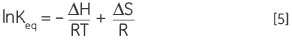
|
Les données de la Figure 1 peuvent servir à déterminer Keq aux températures dans la région de transition. Désignée enthalpie de van't Hoff, l'enthalpie ainsi déterminée dépend de la forme du tracé DSC. Il ne s'agit pas d'une mesure directe de la chaleur de la réaction.
Comme son nom l'indique, la calorimétrie, du latin, calor, qui signifie chaleur, et metrium, qui signifie mesure, constitue un moyen de déterminer directement l'enthalpie dans le cadre de la dénaturation d'une protéine. Il existe des calorimètres hypersensibles, tels que le modèle MicroCal™ VP-Capillary DSC, permettant de mesurer avec précision l'enthalpie à partir d'un fragment de mg de matériau. Ces instruments sont faciles à utiliser, précis et fiables, ce qui fait de ce type de mesures calorimétriques une opération routinière dans tout laboratoire biophysique. Les systèmes fonctionnent en mesurant la capacité calorifique (Cp) d'un échantillon de solution de protéine, tout en balayant les plages de températures. Cp correspond simplement à la quantité d'énergie requise pour augmenter la température de l'échantillon d'une certaine valeur, normalement d'1 K. Elle est liée à l'enthalpie de la loi de Kirchoff :

|
L'excédent de capacité calorifique (différentielle) de la protéine est mesuré par rapport à une cellule de référence à solvant minutieusement dosé lors de l'analyse. Ces instruments sont des calorimètres d'analyse différentielle.
La Figure 1 illustre une mesure par DSC, excepté que la propriété de la protéine étudiée lors de la dénaturation est ici la capacité calorifique. On observe alors une transition de conformation, comme en Figure 2.
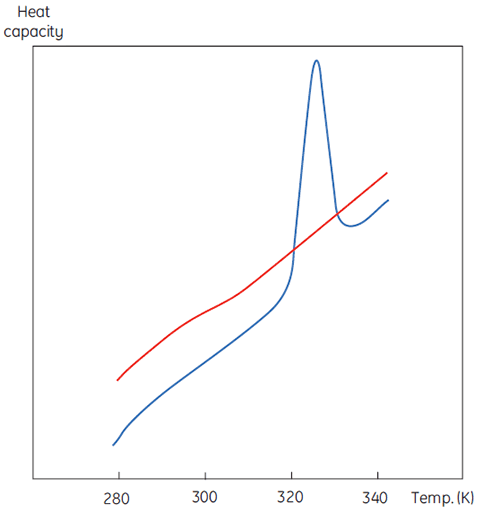
|
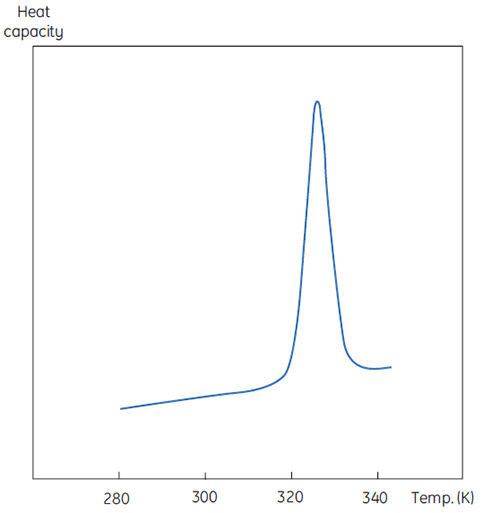
|
Grâce à l'Équation 6, on constate que pour obtenir l'enthalpie, il est nécessaire d'intégrer la fonction d'excédent de capacité calorifique. Pour ce faire, la ligne de base instrumentale observée quand l'échantillon et les cellules de référence contiennent uniquement du solvant doit être exclue. Pour des raisons techniques, la ligne de base instrumentale ne donne pas d'excédent de capacité calorifique entre les cellules (ligne rouge entière, Fig 2, panneau du haut). Après soustraction de la ligne de base, les régions linéaires des deux côtés du pic de transition, représentant la capacité calorifique des états natif et dénaturé de la protéine, sont extrapolés dans la transition, puis fusionnés conformément à la progression de la transition. Cette opération s'effectue à l'aide de routines logicielles. Ensuite, la zone sous le pic obtenu est intégrée pour obtenir l'excédent énergétique requis par la DSC pour dénaturer la protéine dans la cellule d'échantillon. À condition que la concentration de la solution de protéine et le volume de la cellule du calorimètre soient connus, cette énergie peut être convertie en ΔH, exprimée en calories ou joules par mole de protéine. Normalement, la valeur ΔHcal permet d'indiquer une enthalpie calorimétrique directement mesurée.
Le MicroCal VP-Capillary DSC est un instrument très sensible, capable de mesurer des changements infimes de capacité calorifique que l'on associe avec la dénaturation des protéines dans une solution diluée. La capacité calorifique du solvant dépasse celle de la protéine de plusieurs ordres de grandeur. Par conséquent, pour les mesures les plus précises et fiables, il faut minutieusement éliminer la capacité calorifique en bruit de fond, en veillant à ce que le solvant de l'échantillon de protéine et des solutions de référence présentent une composition rigoureusement identique. Dans cette optique, la dialyse et la chromatographie constituent des méthodes adéquates. Le dialysat final ou le flux de colonne doit alors servir de solution de référence.
Les données en Figure 2 peuvent également être analysées comme toute autre propriété de la protéine subissant un changement de dénaturation, conformément aux explications précédentes. Cette approche permet d'obtenir la valeur Tm et l'enthalpie de van't Hoff dépendant d'un modèle. On parle désormais de ΔHvH, pour la distinguer de ΔHcal. La comparaison entre ces deux mesures d'enthalpie, obtenues dans la même expérience, constitue un test utile pour le modèle de dénaturation de protéine. ΔHvH indique l'énergie par mole de l'unité coopérative à l'équilibre, tandis que ΔHcal indique l'énergie par mole de protéine. Si ces énergies sont identiques, la protéine et l'unité coopérative sont égales, ou peuvent être considérées comme présentant une masse molaire identique, ce qui confirme la supposition d'équilibre à deux états.
Dans d'autres scénarios, ΔHvH est inférieure à ΔHcal, ce qui suggère que la masse molaire moyenne de l'espèce à l'équilibre est inférieure à celle de la protéine et qu'un processus de dénaturation impliquant des intermédiaires (I) pourrait s'avérer plus adéquat :

|
Dans des cas extrêmes d'intermédiaires qui se produisent lors de la dénaturation, notamment quand des protéines présentent des domaines de repliement indépendants pour différentes stabilités thermiques, on peut observer deux transitions distinctes. Dans ce cas, la valeur ΔHvH de chaque transition reflète la masse molaire de chaque domaine, tandis que ΔHcal, qui dépend de la masse molaire de la protéine entière, reflète l'énergie totale requise pour dénaturer le système, c'est-à-dire la zone totale pour les deux transitions.
Dans certains cas, ΔHvH est supérieure à ΔHcal, ce qui suggère que la taille ou la masse molaire de l'espèce à l'équilibre est supérieure à celle de la protéine. Cette situation se présente quand la protéine forme des dimères, tétramères ou des agrégats d'ordre supérieur, dont le rapport de ΔHvH sur ΔHcal reflète, tout au moins en principe, l'ordre (n) d'association :

|
Contrairement à l'Équation 7, il s'agit toujours d'un équilibre à deux états, puisque N et D sont inclus. La différence de rapport de ΔHvH sur ΔHcal reflète le fait que le terme de concentration par mole dans le calcul s'appuie sur la sous-unité, et non pas sur la protéine oligomérique.
Ce phénomène révèle également une observation importante, à savoir que la valeur de ΔHcal dépend entièrement de la concentration de l'échantillon. Toute erreur de concentration se retrouvera directement dans l'estimation de l'enthalpie. Il peut notamment s'agir d'erreurs dans la mesure physique de la concentration, habituellement par spectroscopie d'absorbance, d'erreurs dans le coefficient d'extinction de la protéine, ainsi que d'erreurs causées par l'utilisation de matériaux d'une pureté inférieure à 100 % (les protéines contaminantes contribuent à l'absorbance, mais présentent des valeurs Tm différentes, qui pourraient ne pas être détectées par DSC) ou des matériaux natifs au repliement inférieur à 100 % (la protéine dénaturée contribue à l'absorbance, mais elle est déjà dépliée). Il est donc nécessaire d'interpréter avec prudence les variations plus modestes du rapport de ΔHvH sur ΔHcal.
La quantification précise de la pureté et de la concentration de l'échantillon représente l'élément le plus important dans la conception expérimentale et l'interprétation des données pour la DSC.
Si le rapport de ΔHvH sur ΔHcal suggère un schéma de dénaturation oligomérique (Éq. 8), il est facile de confirmer cette hypothèse en vérifiant la dépendance à la concentration de la mesure par DSC. Dans la mesure où l'équilibre implique un changement de concentration de la forme monomérique, la concentration du monomère affectera la position d'équilibre, par action de masse ou en vertu du principe de Le Chatelier. Il est donc probable que la stabilité d'un système oligomérique augmente de concert avec la concentration, comme on peut l'observer dans la pratique (1,2).
L'un des tests les plus importants dans le cadre des mesures par DSC consiste à vérifier la concentration de l'échantillon. Il est toujours avisé de procéder à cette étape si la disponibilité de l'échantillon le permet, même quand le rapport de ΔHvH sur ΔHcal est proche de l'unité.
Après avoir effectué une mesure par DSC, on peut effectuer une expérience simple et potentiellement riche en enseignement, consistant à analyser une nouvelle fois le même échantillon. Quand on observe un endotherme identique lors de cette nouvelle analyse, on peut conclure que l'interaction est totalement répétable avec les états natif et dénaturé à l'équilibre. Cependant, dans un grand nombre de cas, il n'existe pas de transition, ou la transition est d'une envergure réduite lors d'une nouvelle analyse de la protéine. Cela indique qu'une étape irréversible s'est produite lors du mécanisme de dénaturation :

|
Dans ce schéma unimoléculaire (U), des processus tels que la désamidation ou l'isomérisation de la proline peuvent entraîner des modifications irréversibles et empêcher le repliement. Il est souvent possible d'améliorer la répétabilité en effectuant les analyses à des températures légèrement supérieures à la région de transition, et non pas à la température maximale pour la DSC, car l'étape irréversible comporte une composante cinétique (taux). Il est probable que ce taux soit plus élevé à des températures supérieures. Ainsi, plus la température est élevée et la période en état dénaturée longue, plus l'étape irréversible progresse.
L'étape irréversible peut également provenir de l'association ou l'agrégation (Ag) de l'état dénaturé :
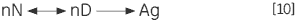
|
Ce phénomène se manifeste par une dépendance à la concentration pour les paramètres d'analyse initiale. Lorsqu'on atteint des volumes de concentration plus élevés, la concentration de D augmente, provoquant une accélération de l'étape irréversible et donc une baisse de Tm pour la protéine. En cas d'agrégation importante et rapide, il peut même s'avérer possible de déformer et tronquer la transition de dénaturation avec l'agrégation exothermique et d'observer des données bruitées au-dessus de la région de transition, comme dans la figure ci-dessous.
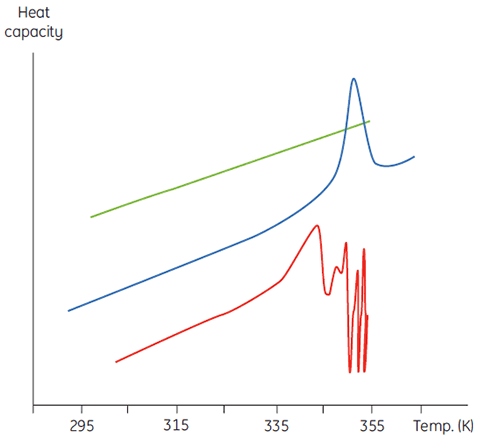
|
À ce titre, la calorimétrie constitue une technique ne souffrant d'aucun compromis. Souvent, la DSC signale des problèmes d'agrégation peu évidents lors d'expériences spectroscopiques de dénaturation thermique effectuées à des concentrations d'échantillon similaires. Dans les deux cas d'irréversibilité, il s'avère instructif d'examiner la concentration de l'échantillon et la dépendance au taux d'analyse pour les mesures par DSC. Justement, dans certains cas, ce type d'informations permet d'extraire des paramètres significatifs à partir de l'établissement de l'équilibre initial et même d'estimer les valeurs des taux irréversibles (3,4).
La recherche d'une dépendance au taux d'analyse dans les mesures DSC constitue un test clé du mécanisme et s'avère toujours intéressante si la disponibilité d'échantillon le permet, y compris quand le système est hautement répétable et à l'équilibre. Même pour les équilibres unimoléculaires les plus simples (Éq. 1), il convient de confirmer que les concentrations d'équilibre en N et D reflètent la stabilité du système dans ces conditions. Ces concentrations évoluent à un taux égal à la somme de la réaction vers l'avant (dénaturation) et l'arrière (repliement) à l'équilibre. Ainsi, lorsque la température augmente plus vite que le système ne peut changer, la position et la forme de l'endotherme DSC subissent une déformation. Ce phénomène donne lieu à des valeurs Tm inexactes et à des rapports anormaux pour ΔHvHcal sur ΔHcal. On considère en général que 60 K/h constitue un bon point de départ pour la majorité des protéines.
En plus de cette utilisation comparative des enthalpies, la technique permet de déterminer avec précision l'enthalpie et la valeur Tm. Comme nous l'avons déjà évoqué, Tm reflète la stabilité thermique de la protéine. Il ne faudrait pas considérer qu'il s'agit d'une équivalence directe avec la stabilité au sens de la longévité de l'activité dans l'échantillon ou de sa durée de vie, puisque ce paramètre peut s'accompagner de l'ajout d'une dimension cinétique supplémentaire par une étape irréversible. Dans l'équilibre le plus simple (Éq. 1), avec réversibilité parfaite, la protéine dispose en théorie d'une durée de vie infinie. Évidemment, à une température supérieure à Tm, l'état natif et actif est moins répandu, ce qui entraîne une baisse d'activité correspondante. Sous Tm, le système peut potentiellement reprendre une structure d'état natif. Dans le cas le plus simple de dénaturation irréversible (Éq. 9), aux températures supérieures à Tm, le taux d'étape en non-équilibre, qui rivalise avec le taux de repliement de D à N, détermine la période pendant laquelle la protéine aura la possibilité de remplir l'état actif et natif. Même bien en deçà de Tm, quand la constante d'équilibre place la majeure partie de la protéine en état natif, en raison du caractère irréversible de la phase D à U, les molécules ne sont plus en état d'équilibre après conversion en U. Pour conserver le rapport [D]/[N] adéquat, conformément à Keq, la protéine native subit une dénaturation. Là encore, le taux cinétique détermine la durée de vie de la protéine native et active. Cela étant dit, il est clairement judicieux de conserver une protéine à un seuil nettement inférieur à sa valeur Tm. Le bon sens suggère qu'il existe un lien entre Tm et la durée de vie. Tous les taux de réaction doivent être ralentis aux températures inférieures, notamment lors de l'étape de D à U. Par ailleurs, comme c'est souvent le cas, l'étape irréversible implique l'agrégation d'au moins deux protéines dénaturées (Éq. 10). Plus la concentration de D est basse, plus le taux sera lent pour cette réaction.
Il est également erroné d'établir une comparaison entre la stabilité thermique et la stabilité à l'équilibre d'une protéine aux températures éloignées de sa valeur Tm. L'extrapolation de la constante d'équilibre, et par extension, de ΔG, au-delà de Tm est une opération complexe, dépendant d'un certain nombre de paramètres évoqués ci-dessous. Cette extrapolation est singulièrement non-linéaire, de sorte que, pour les températures basses, une protéine possédant une valeur Tm élevée n'affichera pas forcément un ΔG supérieur à celui des protéines pour qui la valeur Tm est plus basse.
Les enthalpies déterminées par une expérience de DSC concernent la valeur Tm de mesure, puisqu'il s'agit du point médian de leur détermination. À cette température, ΔG est égale à 0, et l'Équation 4 permet d'établir que ΔS = ΔH / Tm. En d'autres termes, ΔS s'obtient par un processus d'élimination, mais pas par le biais d'une mesure. Toute erreur dans la détermination de ΔH et de Tm sera propagée à ΔS.
Idéalement, on pourrait espérer que ces quantités thermodynamiques fondamentales permettent de glaner des enseignements profonds concernant les forces de stabilisation des structures de protéines. Après tout, les protéines peuvent s'apparenter à des pièces de Lego™ biologique. En ce qui concerne ces jouets, les différents types de briques sont en nombre restreint et elles s'emboîtent selon des permutations limitées. Pourtant, elles donnent lieu à une immense diversité de structures et fonctions dans le cadre de ces paramètres. En biologie, il n'existe qu'un nombre limité de groupes chimiques (les briques) pouvant entrer en interaction dans un polypeptide de protéine. Il peut s'agir d'un groupe du carboxyle et de l'amide de chaîne et des chaînes latérales d'acides aminés. Ces groupes exploitent un ensemble tout aussi réduit d'interactions pour forger des liens : forces de Van der Waals, liaisons hydrogène, charges électrostatiques-attractions de charge et effets hydrophobes. En raison de leur nature non covalente, ces interactions constituent des systèmes à l'équilibre entre deux extrêmes, l'interaction et la non interaction, ce qui permet de les décrire selon les observations thermodynamiques énoncées ci-dessus. Ainsi, les valeurs ΔH et ΔS déterminées pour la dénaturation d'une protéine doivent permettre d'en savoir plus sur la nature et l'intensité de ces interactions.
Malheureusement, il s'agit là de quantités riches en informations. Dans une protéine moyenne, il existe des centaines d'interactions qui s'ajouteront à l'échelle de la molécule entière afin de déterminer ses propriétés thermodynamiques. Il est impossible d'intervenir sur une interaction en l'isolant. En règle générale, la protéine affiche un haut degré de coopération dans sa structure, de sorte qu'il existe deux cas de figure : soit toutes les interactions sont effectives et la protéine est native, soit toutes les interactions sont rompues et la protéine est dénaturée.
Par ailleurs, chaque groupe chimique de la protéine est capable d'interactions intermoléculaires avec l'eau et les sels dissous et intramoléculaires, ainsi qu'avec ses homologues. Les propriétés thermodynamiques de chaque interaction reflètent donc la différence nette entre les groupes qui se rapprochent et ceux qui sont solvatés séparément par des molécules d'eau, ce qui complique encore plus la situation.
Bien qu'il soit impossible d'interpréter les valeurs ΔH et ΔS indépendamment, il existe bien d'autres moyens d'utiliser ces paramètres. Ils sont déterminés à Tm où ΔG est égale à 0. Par conséquent, en connaissant leur dépendance envers la température, il est possible de calculer ΔG à toutes les autres températures. Pour ce faire, il faut connaître le changement de capacité calorifique (ΔCp) pour la dénaturation de la protéine. Puisque la DSC mesure la capacité calorifique, le changement de dénaturation peut être potentiellement déterminé à partir de ces données (Fig 2). À ce titre, la fonction de ligne de base de progression doit être appliquée avant l'intégration de la zone de pic en raison de cette différence.
ΔCp pour les protéines est, comme l'indique la Figure 2, élevée et positive. Sa valeur peut s'obtenir à partir de toute expérience par DSC, mais il est également possible de la déterminer avec autant de précision à partir de la taille de la protéine (5). Plus la protéine est grosse, plus ΔCp est élevée, ce qui reflète le fait que sa valeur est clairement liée à la surface non polaire enfouie par la protéine quand elle se replie en état natif.
La variation de ΔH et de ΔS en fonction de la température représente une autre manière d'exprimer la relation de Kirchoff :
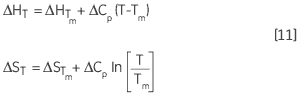
|
ce qui permet d'obtenir une dépendance de ΔG envers la température, calculée selon :
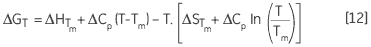
|
L'Équation 11 fournit également une autre manière de déterminer ΔCp. Si la valeur ΔH d'une protéine se mesure dans des conditions de stabilité différente, la variation de ΔH, δΔH/δT, donnera ΔCp. À condition que ces expériences utilisent des tampons adéquats comme nous l'énoncerons ci-après, il s'agit probablement de la manière la plus précise d'obtenir ΔCp, puisque cette méthode repose sur un certain nombre de mesures indépendantes de ΔH.
Grâce à l'Équation 12, on peut calculer ΔG, ΔH et ΔS à toute autre température voulue. Ce genre d'estimation comprendra une erreur reflétant l'erreur originale dans les valeurs Tm et ΔH mesurées, ainsi que l'envergure de l'extrapolation de température au-delà de Tm. En ce qui concerne les extrapolations de température plus longues, les erreurs cumulées peuvent atteindre un seuil considérable et il convient de proposer une estimation de leur envergure en les mentionnant. Pour comparer ΔG, ΔH et ΔS d'un système à l'autre ou dans différentes conditions de solvant, il s'avère essentiel d'extrapoler les données mesurées vers une température commune de comparaison.
Il n'est pas pertinent de comparer les ΔH de dénaturation déterminées par le biais d'expériences pour deux protéines, puisqu'elles sont mesurées à leur seuil Tm respectif. Une telle comparaison nécessite une extrapolation des données ΔH vers une température commune à l'aide du ΔCp de chaque protéine. Cette température correspond de préférence au point médian entre les deux valeurs Tm (T1m+ T2m)/2, afin de minimiser les erreurs d'extrapolation.
Sur le plan qualitatif, il est possible d'établir des comparaisons de ce genre, ce qui permet parfois de révéler un changement brut dans le système. Comme nous l'avons déjà vu, la complexité du processus de dénaturation complique considérablement toute interprétation des données ou des changements de leur valeur. Néanmoins, il existe des stratégies expérimentales qui pourraient à terme permettre de comprendre les contributions d'interactions spécifiques dans la stabilisation de structures de protéines. De par sa capacité à modifier les chaînes latérales d'une protéine, une mutagénèse localisée se prête à un usage très subtil, par exemple dans le cadre de la suppression d'un fragment de méthyle enfoui dans le cœur de la protéine.
Si les propriétés thermodynamiques de ces mutations peuvent être amplifiées à l'aide d'informations structurelles en haute résolution sur la protéine parente et la mutante, cela permet d'envisager une interprétation des changements. Il est possible d'étendre ce type d'approche à des cycles de mutation doubles ou triples, afin de confirmer la validité des énergies d'interaction ou d'inclure les changements de solvant (H20 par rapport à D20) pour étudier les liaisons hydrogène (6).
Nous avons donc établi que la complexité et la teneur des données thermodynamique s'avèrent problématiques. Cependant, il existe des moyens d'exploiter ces caractéristiques, ce qui confère à la DSC des applications très utiles. Ainsi, les chaînes latérales d'acides aminés dans les protéines présentant des groupes protonatables peuvent subir un changement de pKa lors de la dénaturation. En état natif, elles entrent en interaction avec d'autres groupes chargés, tandis que dans la molécule dénaturée, elles entrent en interaction avec le solvant. Le pKa est simplement une représentation de la constante d'équilibre pour le groupe, entre ses formes protonée et non protonée. Si cet équilibre vient à changer lors de la dénaturation, des protons sont libérés ou absorbés par la protéine. Dans la mesure où un tampon présent dans les études préserve le pH (concentration en protons), les protons participant à l'équilibre de dénaturation de la protéine sont récupérés ou libérés par le tampon. Ce processus bénéficie d'une enthalpie correspondante. Cette enthalpie diffère selon les tampons en fonction des caractéristiques chimiques. Par conséquent, si l'on mesure ΔH pour la dénaturation de protéines avec différents tampons et au même pH, on obtiendra différentes valeurs, ce qui reflète la somme de ΔH pour la protéine et ΔH pour l'ionisation de tampon. En représentant sous forme graphique les valeurs observées en comparaison avec la valeur ΔH d'ionisation pour les tampons, on obtient une fonction linéaire dont la pente (positive ou négative) reflète le nombre de protons (somme de tous les décalages pKa) associés à la dénaturation de la protéine.
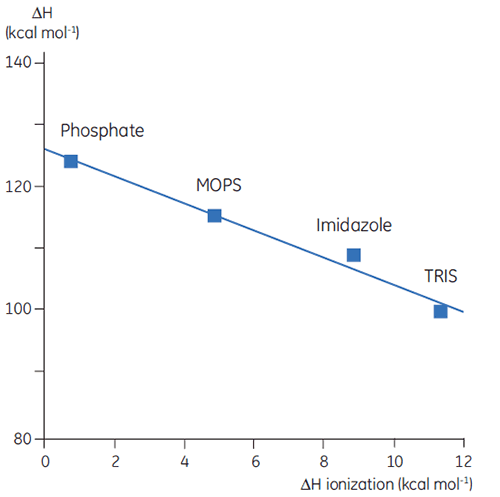
|
Le choix du tampon revêt une grande importance lors de la conception d'expériences en DSC. Pour les tâches routinières, un tampon présentant une enthalpie d'ionisation modeste ou nulle s'avère préférable car cela élimine toute contribution d'ionisation lors de la mesure. Les tampons tels que le formiate, l'acétate et le phosphate constituent des choix avisés, car ils présentent des chaleurs d'ionisation négligeables, sont disponibles sous forme hautement purifiée et sont peu coûteux. Par ailleurs, ces tampons présentent un avantage supplémentaire : leur pH reste constant, même lorsque la température évolue, ce qui constitue un facteur important dans les mesures par DSC. Ce phénomène provient lui aussi de leur faible chaleur d'ionisation.
Autre corolaire d'une protéine possédant des groupes protonatables dont la valeur pKa diffère aux états natif et dénaturé : ses valeurs Keq, et donc ΔG, dépendent de la concentration en protons de la solution, à savoir le pH. Ces valeurs découlent de la loi d'action de masse ou du principe de Le Chatelier. Si la dénaturation Keq affecte le pH de la solution en libérant ou en attirant des protons, le pH de la solution affectera la valeur Keq de la protéine. On peut quantifier cet effet sous la forme suivante :
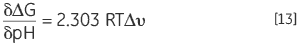
|
où Δυ désigne le changement lors de la protonation. Plus le changement de protonation lors de la dénaturation est élevé, plus le changement de stabilité pour chaque unité de pH sera important. Cet effet ne se manifeste que lorsqu'on observe une différence potentielle d'état de protonation entre le pKa du groupe dans la solution, exposé en état dénaturé, et le pKa du groupe en état natif où il est susceptible d'entrer en interaction avec des groupes de charge opposée, engendrant une baisse de son pKa, ou avec des groupes de charge similaire, provoquant une augmentation de son pKa. Les changements de pKa habituels dans les protéines peuvent atteindre deux unités. Les protéines présentent normalement un profil de pH de stabilité en forme de cloche, comme l'illustre parfaitement cet effet. Le pKa des groupes acides dans les protéines se situe aux environs de 4, tandis que celui des groupes basiques avoisine 10. Les changements de stabilité principaux ont lieu entre 2 et 6 et entre 8 et 12. À un pH neutre, la seule valeur pKa est celle de l'histidine, ce qui peut éventuellement modifier la stabilité dans cette région.
La DSC permet également de déterminer la stabilité thermique d'une protéine à quasiment tout pH étudié. Les valeurs Tm et ΔH peuvent servir à calculer ΔG à une température de comparaison fréquente. Le profil de stabilité de pH peut être analysé à l'aide de l'Équation 13 pour obtenir le changement de protonation pour la dénaturation à tout pH. Les effets d'une mutation spécifique des résidus chargés dans la protéine pour ces profils peuvent indiquer quels groupes participent au comportement de protonation.
Il peut s'avérer utile de considérer les protéines comme de simples ligands reliés par des liaisons non covalentes, présentant différentes « affinités » (pKa) pour les états natif et dénaturé. En tenant compte de cette analogie, nous pouvons immédiatement observer que les effets d'action de masse doivent s'appliquer à tout ligand présentant une affinité de liaison différente des états natif et dénaturé. La plupart, voire l'intégralité, des ligands présentant un intérêt biologique forgent des liaisons étroites, spécifiques à l'état natif de leurs protéines apparentées et n'ont aucune affinité avec l'état dénaturé correspondant. Par conséquent, lorsqu'elles se dénaturent, elles libèrent un ligand dans la solution, ce qui affecte la concentration. L'effet d'action de masse concerne un changement de concentration de ligands dans la solution, qui affecte l'équilibre natif-dénaturé, c'est-à-dire la stabilité de la protéine.
Cette approche peut se décliner de deux manières, tout d'abord en tant qu'outil de recherche brute de ligands qui se rattache à une protéine. La stabilité thermique d'une protéine se mesure indépendamment, puis en présence des ligands à analyser. Ils sont ajoutés à certains niveaux de concentration excessive à la protéine pour garantir que les ligands présentant des affinités raisonnables provoqueront une saturation des sites de liaison sur la protéine. Lorsque la stabilité thermique augmente, il semblerait qu'à la température de dénaturation, une stabilisation de la protéine se produit par le biais d'une liaison à l'état natif. Selon un principe inverse, toute baisse de la stabilité thermique suggère une liaison de ligands à l'état dénaturé mais pas à l'état natif. Il importe de faire preuve d'une certaine circonspection si les changements de stabilité thermiques sont modestes, car les ligands pourraient également intervenir de manière indirecte en modifiant la force ionique ou le pH, ce qui est susceptible de modifier la stabilité. Divers contrôles permettent de résoudre ces éventualités.
Une seconde méthode plus quantitative de cette méthode consiste à mesurer la stabilité thermique en tant que fonction de concentration croissante en ligands. On obtient des données comme celles de la Figure 5, ce qui permet d'atteindre une estimation de la constante de liaison entre le ligand et protéine (7).
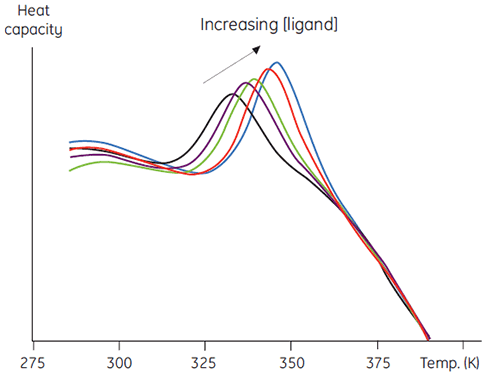
|
Cette méthode présente divers avantages par rapport à des méthodes plus conventionnelles. La méthode permet entre autres de déterminer des constantes de liaison très intenses, impossibles à mesurer par des méthodes d'équilibre conventionnelles. Le signal mesuré est ΔH, une caractéristique universelle de la dénaturation d'une protéine. Il est donc inutile de concevoir des méthodes spécifiques pour chaque protéine étudiée. Par ailleurs, il est possible d'utiliser des concentrations en ligands élevées, qui peuvent s'avérer problématiques dans les méthodes spectroscopiques. Des mélanges de ligands permettent de rechercher des liaisons compétitives à un ligand connu ou d'utiliser des protocoles d'analyse par combinaison complexes.
Les avancées récentes dans l'instrumentation et l'automatisation de la DSC ont permis la création d'un modèle DSC, MicroCal VP-Capillary DSC automatisé à haut débit (8). En plus de proposer des chargements automatisés d'échantillons, ces instruments effectuent leurs analyses plus rapidement, ce qui accélère les opérations de mesure. Avant d'utiliser des taux d'analyse plus élevés, il est judicieux de vérifier la dépendance de Tm et des enthalpies envers ce paramètre, pour garantir l'équilibre du système, comme nous l'avons expliqué précédemment. Une fois ce point établi, le système peut effectuer jusqu'à 25 analyses en 24 heures. Il permet le chargement d'échantillons, entreposés dans des compartiments réfrigérés, garantissant un fonctionnement continu pendant une semaine.
Cette approche automatisée de la DSC constitue un outil analytique puissant permettant de caractériser la stabilité des protéines et d'autres biomolécules. Les conditions de solution (pH, force ionique, additifs) affectant la stabilité thermique d'une protéine peuvent être rapidement évaluées. Ces informations s'avèrent parfois utiles pour déterminer les conditions idéales pour des études de durée de vie ou des essais de cristallisation. De même, les ligands qui s'attachent à une protéine, augmentant ainsi sa stabilité thermique, peuvent être caractérisés ou évalués en détail suivant leur identification sur un écran primaire de débit supérieur.
Ce Livre blanc a été rédigé par le Dr Chris M. Johnsson, travaillant actuellement au MRC Centre for Protein Engineering, Hills Road, Cambridge, CB2 2QH, Royaume-Uni. email : cmj@mrc-lmb.cam.ac.uk
Neet, K.E. et Timm, D.E. Conformational stability of dimeric proteins: Quantitative studies by equilibrium denaturation. Prot. Sci. 3, 2167-2174 (1994).
Johnson, C.R. et al. Thermodynamic analysis of the structural stability of the tetrameric oligomerization domain of p53 tumour suppressor. Biochemistry 34, 5309-5316 (1995).
Freire, E. et al. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Ann. Rev. Biophys. Chem. 19, 159-188 (1990).
Lepock, J.R., et al. Influence of transition rates and scan rate on kinetic simulations of differential scanning calorimetry profiles of reversible and irreversible protein denaturation. Biochemistry 31, 12706-12712 (1992).
Myers, J.K. et al. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Prot. Sci. 10, 2138-2148 (1995)
Connelly, P.R. et al. Enthalpy of hydrogen bond formation in a protein ligand binding reaction. Proc. Natl. Acad. Sci. USA, 91, 1964-1968 (1994).
Brandts, J.F. and Lin, L.N. Study of strong to ultratight protein interactions using differential scanning calorimetry. Biochemistry 29, 6927-6940 (1990).
Plotnikov, V.V. et al. An autosampling differential scanning calorimeter instrument for studying molecular interactions. Assay Drug Dev. Technol. 1, 83-90 (2002).
Cooper, A. et al. Differential scanning microcalorimetr, in Protein-Ligand Interactions: hydrodynamics and calorimetry (Harding, S. E. and Chowdhry, B. Z. eds.), Oxford University Press, pp 287-318 (2000).
Freire, E. Differential Scanning Calorimetry. Methods Mol. Biol. 40, 191-218 (1995).
Privalov, P.L. and Potekhin, S.A. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 131, 4-51 (1986).
Plum, G.E. and Breslauer, K.J. Calorimetry of proteins and nucleic acids. Curr. Opin. Struct. Biol. 5, 682-690 (1995).
Clas, S.D. et al. Differential scanning calorimetry : applications in drug development. Pharm. Sci. Technol. Today 8, 311-320 (1999).
Clausse, D. et al. Morphology characterization of emulsions by differential scanning calorimetry. Adv. Colloid Interface Sci. 117, 59-74 (2005).
Beezer, A.E. et al. Pharmaceutical microcalorimetry: applications to long-term stability studies. Int. J. Pharm. 179, 159-165 (1999).
Jelesarov, I. and Bosshard, H.R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics ofbiomolecular recognition. J. Mol. Recognit. 12, 3-18 (1999).
Plotnikov, V.V. et al. A new ultrasensitive scanning calorimeter. Anal. Biochem. 250, 237-244 (1997).
